studio clinico retrospettivo su 122 casi di tumore della mammella
Pubblicato il 17/10/2014
Titolo
Il Metodo Di Bella (MDB), in uno studio clinico retrospettivo osservazionale su 122 casi di tumore della mammella, ha migliorato la sopravvivenza, la risposta obiettiva e il performance status
AutoreIl Metodo Di Bella (MDB), in uno studio clinico retrospettivo osservazionale su 122 casi di tumore della mammella, ha migliorato la sopravvivenza, la risposta obiettiva e il performance status
Dr. Giuseppe Di Bella
Ente o istituzione di appartenenza
Fondazione Di Bella
Indirizzo per corrispondenza
e-mail : posta@giuseppedibella.it
Telefono 051 239662 ;- 051 230369 ; - Fax 051 2961238
Indirizzo postale Via Marconi N°51 CAP 40122 Bologna – Italia
Parole chiave:
Key words: Metodo Di Bella(DBM), carcinoma mammella, somatostatina/octreotide melatonina, retinoidi, vit. E,D,C ,Bromocriptina Cabergolina inibizione estrogenica
Abstract
Obiettivi: incrementare l’efficacia e diminuire la tossicità nella terapia del cancro.
Metodo: il MDB con MLT, Retinoidi, vitamine E, D3, C, esercita un effetto differenziante citostatico, antiangiogenico, immunomodulante, fattorialmente sinergico potenziando contemporaneamente quelle funzioni che la fisiologia considera essenziali per la vita. Con Somatostatina e/o analoghi, il MDB esercita un effetto antiproliferativo, regolando negativamente il più potente mitogeno (GH), recettorialmente co-espresso e interattivo con la Prolattina, inibita da Cabergolina e/o Bromocriptina . La regolazione negativa del GH si estende direttamente ai fattori di crescita GH-dipendenti. Il MDB, nei carcinomi mammari, prevede l‘inibizione estrogenica e minimali dosaggi apoptotici, non citotossici e non mutageni, di Ciclofosfamide o Oncocarbide, la cui tollerabilità è esaltata dalla MLT e dalle vitamine del MDB.
Risultati: La guarigione completa e stabile di 4 casi, e la rapida regressione della neoplasia in altri 5 casi, solo con MDB (terapia di prima linea), senza intervento chirurgico.
Assenza di recidive nell’impiego come terapia adiuvante del MDB.
Sopravvivenza a 5 anni, nel IV° stadio, del 50 % , ampiamente superiore ai dati della letteratura.
Miglioramento pressoché generalizzato della qualità di vita e assenza di rilevante e/o prolungata tossicità.
Conclusioni: la valorizzazione di evidenze scientifiche ancora sottovalutate, come i molteplici meccanismi d’azione antitumorale della MLT, la regolazione negativa dei massimi mitogeni interattivi: GH – GF(fattori di crescita GH dipendenti) - Prolattina, unitamente all’azione differenziante e omeostatica di retinoidi Vit. E, D3, C e MLT, ha consentito questi risultati.
Aspetto essenziale del meccanismo d’azione sulla risposta clinica, il sinergismo fattoriale di ogni componente del MDB .
Abbreviazioni
ATRA –All Trans Retinoic Acid
DBM – Di Bella’s Method
EGF – Epidermal Growth Factor
EGFR – Epidermal Growth Factor Receptor
ER – Estrogen Receptor
FGF – Fibroblastic Growth Factor
G – Gastrin
GF – Growth Factor
GH – Growth Hormone
GHR – Growth Hormone Receptor
HGF – Hepatocyte Growth Factor,
IGF1-2 – Insulin-like Growth Factor 1-2
IGFR – Insulin-like Growth Factor Receptor
IL8 – Interleuchin 8
MRI – Magnetic Resonance Imaging
MLT – Melatonin
NGF – Nerve Growth Factor
NHL – Non-Hodgkin’s Lymphoma
NOSe – Endothelial Nitric Oxide Synthase
PDGF – Platelet-Derived Growth Factor
PET – Positron Emission Tomography
PG2 – Prostaglandin 2
SST – Somatostatin
SSTR – Somatostatin Receptor
TGF – Transforming Growth Factor
TRK – Tyrosine-kinase
VEGF – Vascular Endothelial Growth Factor
VIP – Vasoactive Intestinal Peptide
Metodo
Principi Attivi [ componenti della terapia prescritta (MDB)] :
- Somatostatina: (14 aminoacidi), 3-4 mg iniettata sottocute di notte, in coincidenza del picco notturno di increzione del GH, e nell’arco di 10 ore mediante un temporizzatore ( per la breve emivita - circa 3 minuti).
- Octreotide: analogo della somatostatina (8 aminoacidi) in formulazione ritardo da 30 mg intramuscolo, ogni 25 giorni, per una completa saturazione, sia recettoriale che temporale, con analoghe proprietà antiproliferative e proapoptotiche della somatostatina a 14 aminoacidi .
- Bromocriptina: compresse da 2,5 mg (1\2 cpr mattino e sera), per l’inibizione della prolattina, potente e ubiquitario ormone mitogeno, recettorialmente coespresso, con il GH, sulle membrane cellulari, e con esso interattivo.
- Cabergolina: 1\2 compressa (2 volte la settimana), potenziatore dell’attività della bromocriptina con emivita nettamente maggiore.
- soluzione vitaminica, secondo la formulazione del prof. Di Bella:
Beta carotene…………………… gr 2
Axeroftolo palmitato … ……....gr 1
Ac trans retinoico ( ATRA)…….. gr 1
Alfatocoferile acetato …………..gr 1000
Un cucchiaio medio (100 mg x Kg di peso corporeo), almeno 15’ prima del pasto, 3 volte al di.
Axeroftolo palmitato … ……....gr 1
Ac trans retinoico ( ATRA)…….. gr 1
Alfatocoferile acetato …………..gr 1000
Un cucchiaio medio (100 mg x Kg di peso corporeo), almeno 15’ prima del pasto, 3 volte al di.
- Diidrotachisterolo: (Vit D3 di sintesi) 10 gocce nello stesso cucchiaio assieme al composto vitaminico per ogni somministrazione ( 30 gocce al dì)
- Melatonina: chimicamente complessata con adenosina (mediante un legame di idrogeno) e glicina, secondo la formulazione del Prof. Di Bella: Melatonina 12%, Adenosina 51%, Glicina 37%
- Inibitore delle aromatasi: una compressa al giorno
- Idrossiurea: compresse da 500 mg (1 - 2 al dì) , oppure
- Ciclofosfamide: compresse da 50 mg (1 - 2 volte al dì )
- Calcio: 1 gr. 2 volte al dì, con l’acido ascorbico.
- Ac ascorbico: 2 gr. insieme al calcio in un bicchiere d’acqua (2 volte al dì durante il pasto)
- Condroitinsolfato solfato: Cps 500 mg (2 volte al dì )
CASISTICA
Risultati:Questo studio clinico osservazionale retrospettivo è stato condotto monitorando per almeno cinque anni (92) casi di carcinomi della mammella curati con MDB. Il monitoraggio ha riguardato tutti gli elementi utili ad elaborare uno studio statistico volto a rappresentare correttamente gli effetti clinici e terapeutici conseguiti (sopravvivenza, risposta obiettiva, performance status).
Dei casi studiati, 82% erano carcinomi duttali infiltranti – 13% lobululari Iniltranti – 5% altro).
Altri trenta casi, sono stati anche stati sottoposti a perizie e certificati dal Tribunale di Lecce (Italia). Questi 30 casi hanno documentato, con MDB, un netto miglioramento obiettivo e della qualità di vita dopo il fallimento di precedenti terapie oncologiche e ottenuto da parte dell’Autorità Giudiziaria, per la documentazione dei risultati ottenuti, l’erogazione gratuita dei farmaci MDB (non prevista in Italia da parte del SSN). In questi casi il magistrato, in base alla perizia giurata di un CTU ( consulente tecnico di ufficio ), ha condannato il servizio sanitario nazionale italiano a erogare completamente, e gratuitamente, il MDB.
Consapevoli dei limiti che la statistica assegna a rappresentazioni per numeri limitati di casi, vi invitiamo a considerare, comunque, che non uno, ma tutti i parametri di raffronto con i trattamenti convenzionali sono stati ampiamente superati (anche di diversi percentili), ma soprattutto, che per la prima volta si è potuto assistere ad una serie di guarigioni complete, e permanenti, senza l’ausilio di precedenti trattamenti, né farmacologici, né radianti, né chirurgici.
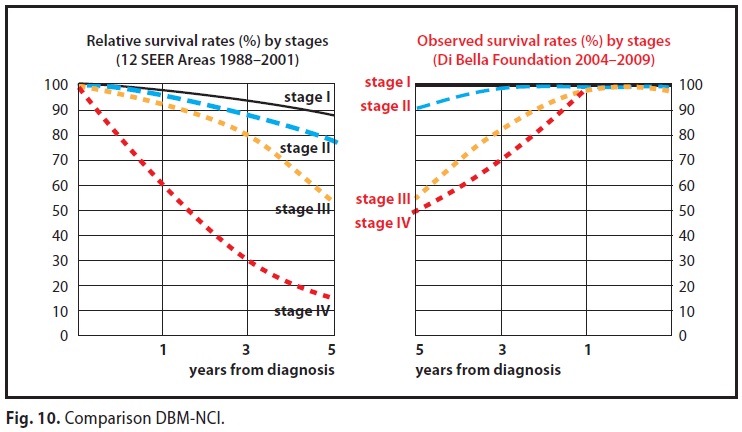
Nella sopravvivenza a cinque anni il MDB ha ottenuto dati nettamente superiori, per ogni stadio, rispetto a quelli che Il National Cancer Institute (N.C.I) ha diffuso attraverso il progetto 12 SEER, Areas
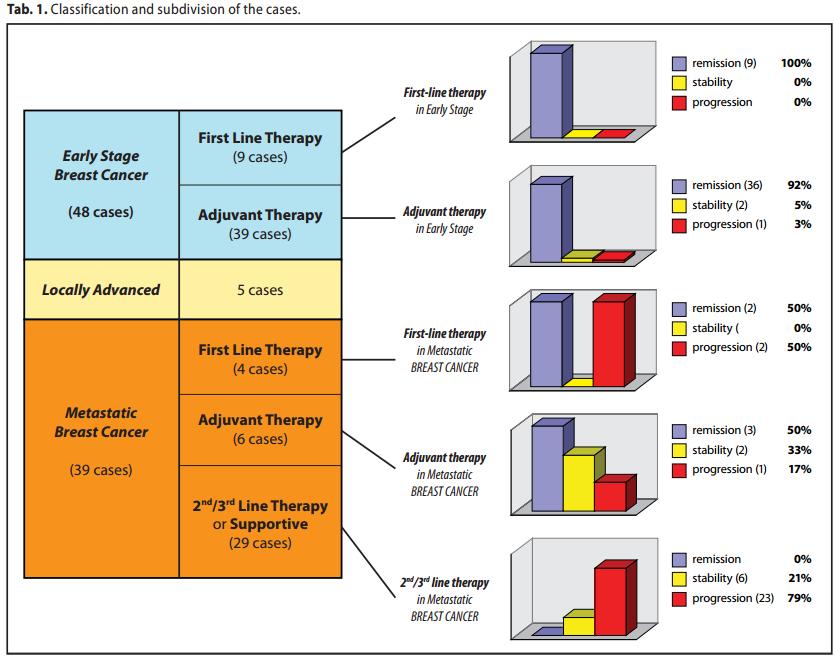
Classificazione e suddivisione dei casi clinici.
Dei 122 casi :
A) 48 erano in stadio iniziale, di essi
A1) 9 hanno effettuato il MDB come terapia di prima linea.
A2) 39 hanno effettuato il MDB come terapia adiuvante.
B) 39 erano in stadio metastatico, di essi A2) 39 hanno effettuato il MDB come terapia adiuvante.
B1) 4 non hanno avuto alcun trattamento medico (MDB in prima linea)
B2) 6 sono stati precedentemente trattati con cchirurgia (MDB come cura adiuvante)
B3) 29 casi sono stati operati e chemio e/o radiotrattati
C) 5 casi hanno iniziato il trattamento in una situazione localmente avanzataB2) 6 sono stati precedentemente trattati con cchirurgia (MDB come cura adiuvante)
B3) 29 casi sono stati operati e chemio e/o radiotrattati
D) 30 casi certificati dal Tribunale di Lecce ( Italia)
per questi casi non si dispone di dati analitici per cui non sono stati ricompresi nelle rappresentazioni statistiche.
EFFICACIA
Tav. 1 - RAPPRESENTAZIONE DEI RISULTATI STATISTICI ( 92 casi )
ANALISI DEI DATI E OSSERVAZIONI
A - Carcinoma mammario allo stadio iniziale ( 48 casi )
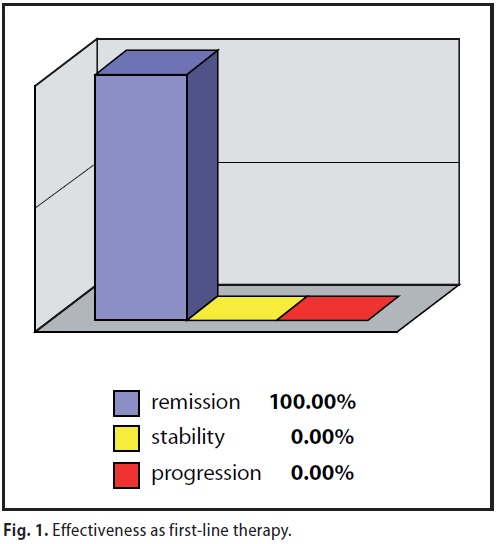
A/1) MDB come Terapia di prima-linea ( 9 casi )
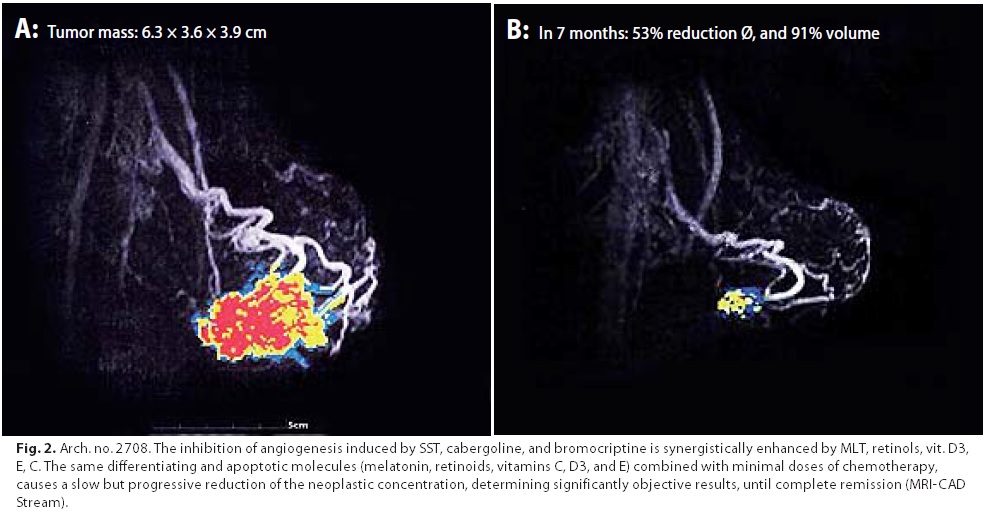
Nove pazienti con carcinoma mammario allo stadio I-II-IIIa hanno rinunciato all’intervento chirurgico scegliendo come unica terapia il MDB. Quattro su nove hanno conseguito una Remissione Completa, tre di esse risultano libere da malattia ormai da oltre 48 mesi , una da 63 mesi (Tabella 2). Le altre cinque sono tuttora in trattamento con progressiva ed evidente riduzione del volume tumorale (Figura 2)
Nove pazienti con carcinoma mammario allo stadio I-II-IIIa hanno rinunciato all’intervento chirurgico scegliendo come unica terapia il MDB. Quattro su nove hanno conseguito una Remissione Completa, tre di esse risultano libere da malattia ormai da oltre 48 mesi , una da 63 mesi (Tabella 2). Le altre cinque sono tuttora in trattamento con progressiva ed evidente riduzione del volume tumorale (Figura 2)
 .
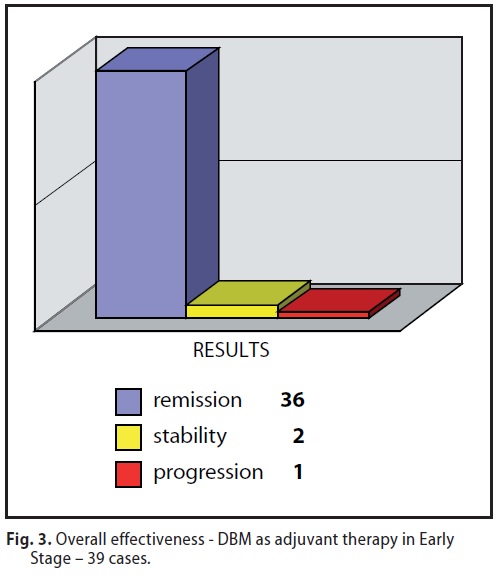
.A/2 ) MDB come Terapia Adiuvante (39 casi) post-chirurgica.
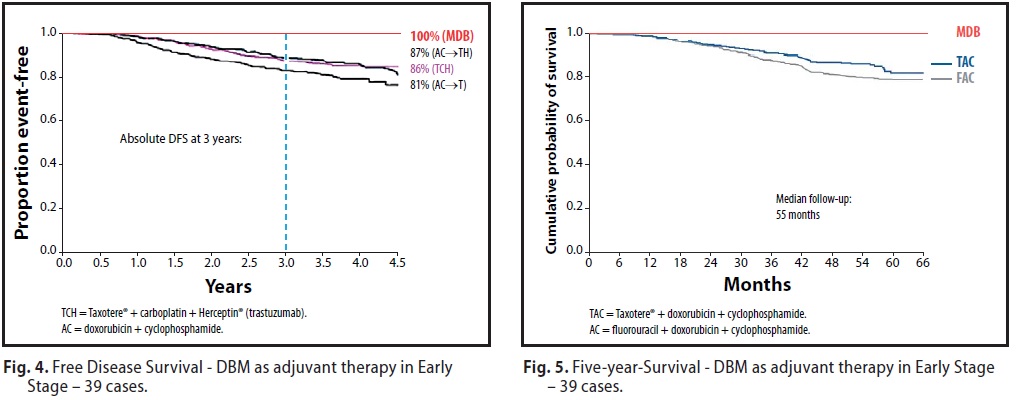
In trentanove casi il MDB è stato impiegato come Terapia Adiuvante con netto e significatico aumento sia della Sopravvivenza Globale (mediana a 60 mesi = 100%) che dell’intervallo libero da malattia.
12 pazienti presentavano, al momento dell’inserimento nel trial terapeutico, evidenti segnali di ripresa della malattia (locale o linfonodale). Relativamente all’efficacia si sono avuti significativi risultati : Le remissioni sono state il 95% (attualmente tutti liberi da malattia) Sopravvivenza a 5 anni: 100%
L’unica progressione si è registrata in una paziente, 2 anni dopo aver interrotto totalmente, improvvisamente, e di propria iniziativa, il trattamento terapeutico 4 (intervallo libero da malattia)
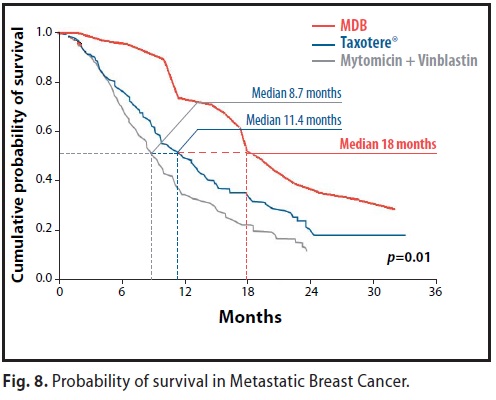
La Fig N° 5 rappresenta il confronto tra i risultati della sopravvivenza del 100% a 5 anni conseguiti dal MDB, con le mediane di sopravvivenza ufficiali ottenute con i seguenti protocolli e combinazioni di chemioterapici:
- TAC = Taxotere + doxorubicina + ciclofosfamide
- FAC = Fluorouracile + doxorubicina + ciclofosfamide
B/1 ) MDB come Terapia di Prima Linea ( senza chirurgia )
4 casi ( 2 remissioni – 2 progressioni )
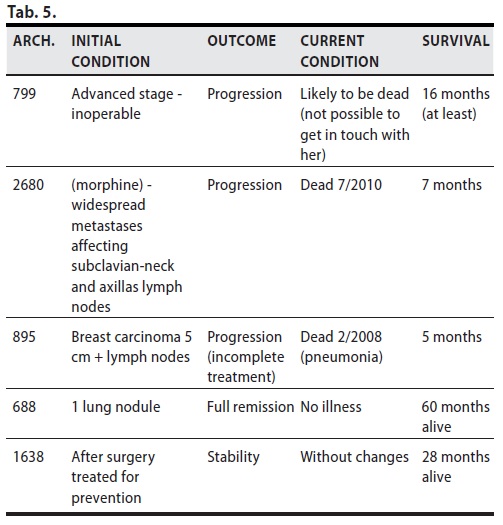
Di esse una paziente con progressione linfonodali , retroperitoneali, mediastiniche e ossee ( Arch 994) ha avuto un’evidente remissione parziale, è vivente e tranne che nelle ossa ha avuto remissione in tutte le altre sedi e buona qualità di vita.
Un’altra (Arch 586) con 2 localizzazioni al seno Dx e metastasi polmonari ha avuto una remissione e a 56 mesi è vivente.
B/2 ) MDB come Terapia Adiuvante
totale 6 casi (3 remissioni – 2 stabili – 1 progressione)
Sei pazienti sono state operate( ma non chemio trattate) e hanno pertanto applicato il MDB come trattamento adiuvante di prima linea.
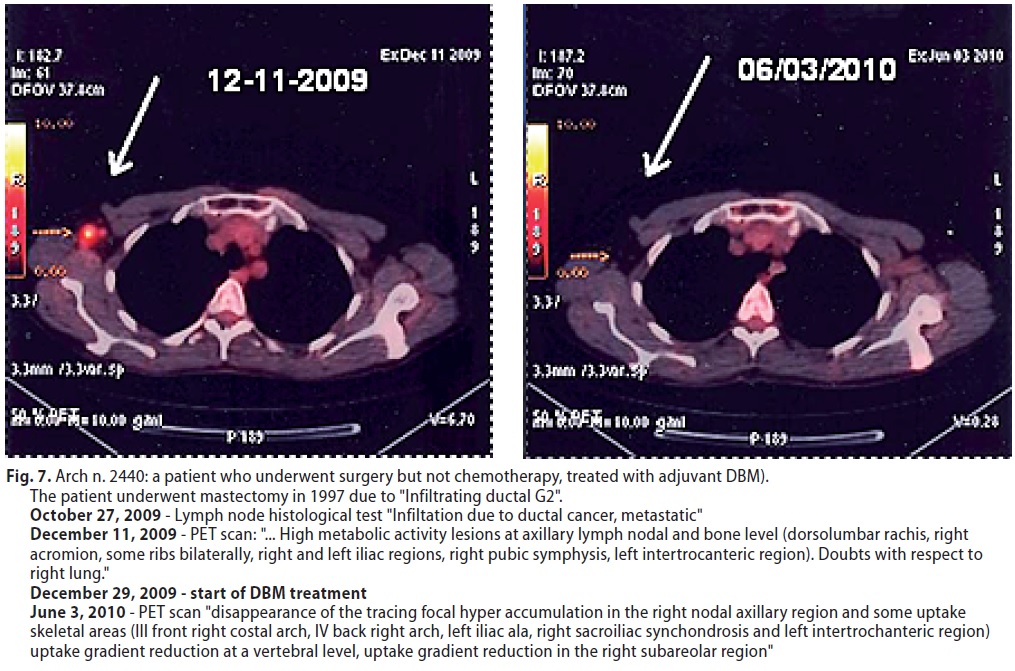
Di esse 5 sono viventi, una con metastasi polmonari e linfonodali( Arch 1826) ha avuto una completa remissione, una con infiltrazione ascellare e ossea disseminata ha avuto una evidentissima remissione parziale ( Arch 2440 - Fig. 7) così come un altro caso con metastasi epatiche ed ossee (Arch 2726). In un caso di metastasi ossee linfonodali e polmonari è stabile, come un caso con metastasi ossee multiple. Un’altra paziente con metastasi in 30 linfonodi su trentadue, e ossee disseminate, è deceduta dopo 38 mesi.
(Fig 7 ° Arch 2440 paziente operata ma non chemiotrattata applicazione del MDB come trattamento di 1° linea )
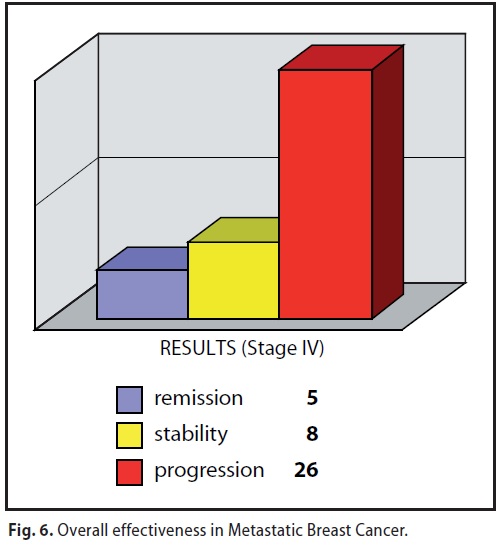
B/3 ) DBM as 2nd /3rd line, or supportive therapy
totale 29 casi (6 stabili – 23 progressioni )
Pazienti sottoposti precedentemente a trattamenti chemioterapici e chirurgici (ora orfani di terapia)
Dati generali: netto e significativo miglioramento della Sopravvivenza Globale
Mediana di sopravvivenza = 18 mesi ( il 30% è vivente a 30 mesi ) rispetto a quelle ottenute nello stesso stadio con vari protocolli chemioterapici
C - Carcinoma mammario localmente avanzato
Nei cinque casi in questione si sono registrati due significativi risultati, la sopravvivenza ad oggi , dopo 60 mesi, con remissione completa, di un voluminoso carcinoma duttale infiltrante ( Arch.688) che al momento dell’inizio del trattamento presentava progressione polmonare, e la stabilità a 28 mesi (Arch. 1638) con sopravvivenza a tutt’oggi di un altro voluminoso carcinoma operato
CONFRONTO CON I DATI DELLE RILEVAZIONI STATISTICHE UFFICIALI
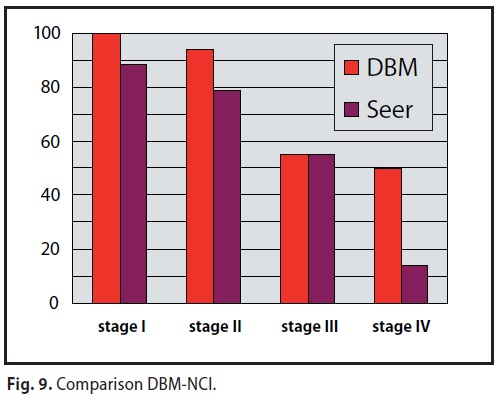
Nel confronto tra MDB e i dati del NCI 12 SEER Areas 1988-2001 relativi alla sopravvivenza a cinque anni, mentre al 3° stadio i dati sono pressoché sovrapponibili, il MDB consegue miglioramenti in ogni altro stadio con scarto particolarmente evidente nel 4° stadio con un 50% rispetto al 14,8 del NCI
RAZIONALE DELLA TERAPIA
Recentemente sono stati ulteriormente approfonditi e confermati i noti meccanismi molecolari del ruolo mitogeno del GH (Milewicz T. 2011; Chiesa 2011) e della Prolattina (Aksamitiene E. 2011) i cui recettori di membrana sono coespressi e interattivi (Chen M. 2011; Breves JP. 2011; Xu J. 2011) così come è stato confermato che i recettori della Somatostatina e Dopamina interagiscono funzionalmente in senso antiproliferativo ( Diakatou 2011; Savenau 2011; Ferone 2010; Gruszka 2001). L’espressione recettoriale ubiquitaria della Prolattina (Sandret L. 2011; Ben-Jonathan N. et al., 2002; Hooghe R. et al., 1998) e del GH (Taboada GF 2010; Lincoln DT et al. 1998; De Souza I. et al., 1974) rappresenta una delle conferme del ruolo mitogeno di queste molecole. Dato confermato anche nei carcinomi della mammella (Wennbo H et al., 2000). È dimostrato anche il rapporto causale e proporzionale tra espressione recettoriale del GH (di cui la SST è l’antitodo biologico) e induzione e progressione tumorale (Friend et al., 2000; Zeitler et al., 2000; Gruszka et al., 2001), rilevando, isto-chimicamente, concentrazioni di GHR nettamente superiori nei tessuti tumorali rispetto a quelli sani. Sono numerose le conferme che l’inibizione del eserciti un effetto antiproliferativo (Bustamante JJ 2010) e, conseguentemente, la somministrazione di GH rappresenti un elevato rischio di induzione neoplastica (Sklar CA 2004). E’ pertanto noto, e ampiamente documentato, il potente ruolo mitogeno del GH, ed il fatto che l’indice proliferativo e la velocità di progressione delle popolazioni neoplastiche risulti direttamente proporzGHionale, in rapporto dose-dipendente, all’espressione recettoriale del GH stesso (Lincoln et al., 1998).
La proliferazione cellulare è anche strettamente dipendente da molecole mitogene GH dipendenti, e da esso positivamente regolate, come EGF, FGF, HGF, IGF1-2, NGF,PDGF, VEGF, TGF, (Yarman 2010; Hagemeister et al., 2008; Taslipinar et al., 2009; Di Bella 2004-2008-2009-2010; Murray et al., 2004; Sall et al., 2004; Szepesházi et al., 1999) oltre che da fattori di crescita prodotti dall’apparato gastrointestinale, come VIP, CCK, PG (Kath et al., 2000).
Sia la proliferazione cellulare fisiologica, che quella neoplastica, avvengono per mezzo di queste stesse molecole, che la cellula neoplastica utilizza in misura esponenziale rispetto a quella sana. Antidoti biologici del GH, come Somatostatina e analoghi, non riducono solo il tasso plasmatico di GH, ma anche l’espressione e la trascrizione di fattori di crescita altamente mitogeni, come IGF1-2 (Cascinu S. et al., 2001; Pollak M. 1997; Schally AV et al., 2001), EGF (Held-Feindt J. et al., 1999), FGF (Mentlein R. et al., 2001), ma estendono la loro regolazione negativa ai rispettivi recettori con evidenti riflessi antiproliferativi e antiangiogenici (Szepesházi K. et al., 1999; Mishima M. et al., 1999; Barrie R. 1993).
E’ noto come l’interazione GH-PRL e l’asse GH-IGF come quello PRL-IGF abbiano una determinata influenza sullo sviluppo biologico neoplastico.
Sta emergendo con evidenza anche l’interazione Estrogeno – IGF, e pertanto la funzione centrale di IGF come modulatore proliferativo comune del GH, PRL, Estrogeno. L’asse estrogeno-IGF è coinvolto nell’iperplasia mammaria, nel carcinoma della mammella ( Klinberg 2010), e interagisce in funzione blastica con altri sistemi endocrini come GH – IGF – PRL – IGF1 (Lynn 2011; Zhou 2011; Mendoza RA et al 2010; Hewitt 2010; Leung 2004; Fürstenberger G. 2003; Juul 2001; Lissoni 1987; Mauri 1985). L’interazione di questi sistemi endocrini mitogeni conferma il razionale MDB che sviluppa l’inibizione contemporanea e sinergica di GH, PRL, Estrogeno e, conseguentemente, di IGF nelle neoplasie mammarie con o senza accertamento dell‘espressione dell’ER. Il nostro risultato clinico sui 122 casi monitorati ha confermato le basi scientifiche e razionali di questa concezione terapeutica. Non solo nei carcinomi mammari, ma in un’elevatissima, subtotale percentuale di varietà di cellule neoplastiche sono stati individuati recettori IGF che rispondono mitogenicamente al ligando. La Somatostatina esercita l’attività antiblastica sia direttamente, attraverso l’inibizione dell’espressione del gene IGF, che indirettamente, mediante la soppressione del GH, da cui dipende l’increzione di IGF (Pollak M. 1997; Schally AV et al., 2001; Schally AV et al., 2003).
Ampiamente documentata è anche l’attività inibitoria della SST su un altro potente fattore di crescita mitogeno, EGF, frequentemente espresso nelle neoplasie mammarie, attraverso molteplici meccanismi:
- inibizione dose dipendente della fosforilazione tirosinica indotta dall’attivazione di EGFR da parte di EGF (Mishima M. et al., 1999);
- riduzione di EGFR nelle cellule tumorali (Szepesházi K. et al., 1999);
- riduzione dell’espressione di EGF (Held-Feindt J. et al., 1999);
- abbattimento della concentrazione plasmatica di EGF (Cascinu S. et al., 2001).
È stato dimostrato che i tumori al seno esprimono SSTR1, SSTR2, SSTR3, meno frequentemente SSTR5 (Albérini J.L. et al., 2000; Schaer J.C. et al., 1997), che almeno nel 50% dei casi sono scintigraficamente visibili, mentre in oltre la metà delle scintigrafie negative, indagini istochimiche hanno rilevato la presenza di STTR. Pertanto la presenza di STTR (Albérini JL et al., 2000; Barnett P et al., 2003; Pinzani P et al., 2001; van Eijck CH et al., 1998), e di recettori neuroendocrini in una rilevante percentuale di questi carcinomi, costituisce un’ulteriore indicazione razionale all’impiego
della SST, peraltro già ampiamente giustificata dalla citata regolazione negativa della SST sul GH, gli oncogeni GH–correlati, e l’angiogenesi.
Promotori dell’angiogenesi, passaggio essenziale della progressione neoplastica, come la cascata dei monociti, l’interleukina 8, la Prostaglandin 2, l’Endothelial Nitric Oxide Synthase, oltre al concorso di fattori di crescita (il cui sinergismo è essenziale per l’angiogenesi stessa), come il VEGF, TGF, IGF1, FGF, HGF,PDGF, sono negativamente regolati da Somatostatina e analoghi (Albini A et al., 1999; Barrie R. et al., 1993; Cascinu S et al., 2001; Florio T et al., 2003; Jia WD et al., 2003; Turner HE et al., 2000; Vidal S et al., 2000; Watson JC et al., 2001; Wiedermann CJ et al., 1993).
L’inibizione dell’angiogenesi indotta dalla SST è sinergicamente potenziata dagli altri componenti del MDB, come la MLT (Lissoni P et al., 2001; Di Bella L et al., 1979; Di Bella L et al., 2006), Retinoidi (McMillan K et al., 1999; Kini AR et al., 2001; Majewski S et al., 1994), vit. D3 (Kisker O et al., 2003; Mantell DJ et al., 2000), Vit. E (Shklar G et al., 1996; Tang FY et al., 2001; Neuzil J et al., 2002), vit. C (Ashino H et al., 2003), inibitori prolattinici (Turner HE et al., 2000), componenti della matrice extracellulare (Liu Y et al., 2005; Ozerdem U et al., 2004) .
Ugualmente l’effetto citostatico, antiproliferativo, antimetastatico della Somatostatina è efficacemente sinergizzato dagli altri componenti del MDB:
- Retinoidi (Hassan HT et al., 1990; Voigt A et al., 2000; Piedrafita FJ et al., 1997; Onogi N et al., 1998)
- MLT (Bartsch C et al., 1999 ; Kvetnoĭ IM et al., 1986 ; Mediavilla MD et al., 1999 ; Maestroni GJ et al., 1996 ; Cos S et al., 2000)
- Vit D3 (Jensen SS et al., 2001 ; Barroga EF et al., 2000 ; Campbell MJ et al., 2000)
- Cabergolina e Bromocriptina [inibitori prolattinici] (Gruszka A et al., 2001; Ben-Jonathan N et al., 2002; Lissoni P et al., 2001; Klijn JG et al., 1996; Manni A et al., 1989)
- Glucosamina solfato, Galattosamina solfato, componenti della matrice extracellulare (Pumphrey CY et al., 2002; Batra RK et al., 1997)
- Vit E (Turley JM et al., 1995; Israel K et al., 2000; Malafa MP et al., 2002; Neuzil J et al., 2002; Shklar G et al., 1996)
- Vit C (Head KA 1998; Murata A et al., 1982; Cameron E et al., 1979)
antiproriliferative e l’ espressione recettoriale di membrana del citosol e nucleare della MLT, unitamente al sinergismo recettoriale con RAR ed RXR dei retinoidi, VDR della vitamina D. Ampie e crescenti sono le conferme dell’intuizione del Prof Luigi Di Bella che aveva definito la MLT condizione necessaria , anche se non sufficiente, nella terapia del cancro (Hill 2011; Korkmaz 2009, Benitez-King 2009; Cabrera 2010; Cucuna 2009; Dong 2010; Girgert Lin 2010; Mao 2010; Mediavilla 2010; Rogelsperger 2009-2010)
Conclusioni:
Questi risultati comprovano la razionalità ed efficacia della concezione multiterapica del MDB che mediante l’interazione sinergica dei suoi componenti asseconda ed esalta le reazioni vitali e l’omeostasi antitumorale per metterle in condizione di contrapporsi alla insorgenza e progressione neoplastica perseguendo :
a) La difesa dall’aggressione neoplastica
b) L’inibizione della proliferazione neoplastica
c) Il contrasto della spiccata tendenza mutagena del fenotipo neoplastico.
Il tumore è deviazione dalla vita normale, per cui occorre riportare le reazioni deviate verso la norma attraverso il potenziamento di tutti quei mezzi che la Fisiologia considera essenziali per la vita.
La documentato sinergismo antiangiogenico di ogni componente del MDB, unitamente a quello antiproliferativo di somatostatina e inibitori prolattinici ed estrogenici, differenziante immunomodulante, trofico e omeostatico degli altri componenti del MDB, hanno conseguito questo risultato rilevante sotto diversi aspetti: la risposta obiettiva completa stabile in diversi pazienti senza intervento, l’incremento particolarmente rilevante nella mediana di sopravvivenza a 5 anni del 4° stadio, un generalizzato e sensibile miglioramento della qualità di vita, effetti collaterali transitori, di modesta intensità assolutamente irrilevanti se confrontati con la nota tossicità della chemioterapia.
REFERENCES
- Aksamitiene E, Achanta S, Kolch W, Kholodenko BN, Hoek JB, Kiyatkin A (2011). Prolactin-stimulated activation of ERK1/2 mitogen-activated protein kinases is controlled by PI3-kinase/Rac/PAK signaling pathway in breast cancer cells. Cell Signal. 23 (11): 1794–805.
- Albérini JL, Meunier B, Denzler B, Devillers A, Tass P, Dazord L, et al . (2000). Somatostatin receptor in breast cancer and axillary nodes: study with scintigraphy, histopathology and receptor autoradiography. Breast Cancer Res Treat. 61 (1): 21–32.
- Albini A, Florio T, Giunciuglio D, Masiello L, Carlone S, Corsaro A, et al. (1999). Somatostatin controls Kaposi’s sarcoma tumor growth through inhibition of angiogenesis. FASEB J. 13 (6): 647–655.
- Ashino H, Shimamura M, Nakajima H, Dombou M, Kawanaka S, Oikawa T, et al. (2003). Novel function of ascorbic acid as an angiostatic factor. Angiogenesis.6 (4): 259–269.
- Barnett P (2003). Somatostatin and somatostatin receptor physiology. Endocrine. 20 (3): 255–264.
- Barrie R, Woltering EA, Hajarizadeh H, Mueller C, Ure T, Fletcher WS (1993). Inhibition of angiogenesis by somatostatin and somatostatin-like compounds is structurally dependent. J Surg Res. 55 (4): 446–450.
- Barroga EF, Kadosawa T, Okumura M, Fujinaga T (2000). Inhibitory effects of 22-oxa-calcitriol and all- trans retinoic acid on the growth of a canine osteosarcoma derived cell-line in vivo and its pulmonary metastasis in vivo. Res Vet Sci. 68 (1): 79–87.
- Bartsch C, Bartsch H, Buchberger A, Stieglitz A, Effenberger-Klein A, Kruse-Jarres JD, et al. (1999). Serial transplants of DMBA-induced mammary tumors in Fischer rats as a model system for human breast cancer. VI. The role of different forms of tumor associated stress for the regulation of pineal melatonin secretion. Oncology. 56 (2): 169–176.
- Batra RK, Olsen JC, Hoganson DK, Caterson B, Boucher RC (1997). Retroviral gene transfer is inhibited by chondroitin sulfate proteoglycans/glycosaminoglycans in malignant pleural effusions. J Biol Chem. 272 (18): 11736–43.
- Benítez-King G, Soto-Vega E, Ramírez-Rodriguez G (2009). Melatonin modulates microfilament phenotypes in epithelial cells: implications for adhesion and inhibition of cancer cell migration. Histol Histopathol. 24 (6): 789–799.
- Ben-Jonathan N, Liby K, McFarland M, Zinger M (2002). Prolactin as an autocrine/paracrine growth factor in human cancer. Trends Endocrinol Metab. 13 (6): 245–250.
- Breves JP, Seale AP, Helms RE, Tipsmark CK, Hirano T, Grau EG (2011). Dynamic gene expression of GH/PRL-family hormone receptors in gill and kidney during freshwater-acclimation of Mozambique tilapia. Comp Biochem Physiol A Mol Integr Physiol. 158 (2): 194–200.
- Cabrera J, Negrín G, Estévez F, Loro J, Reiter RJ, Quintana J (2010). Melatonin decreases cell proliferation and induces melanogenesis in human melanoma SK-MEL-1 cells. J Pineal Res. 49 (1): 45–54.
- Cameron E, Pauling L, Leibovitz B (1979). Ascorbic acid and cancer: a review. Cancer Res. 39 (3): 663–681.
- Campbell MJ, Gombart AF, Kwok SH, Park S, Koeffler HP (2000). The anti-proliferative effects of 1alpha,25(OH)2D3 on breast and prostate cancer cells are associated with induction of BRCA1 gene expression. Oncogene. 19 (44): 5091–5097.
- Cascinu S, Del Ferro E, Ligi M, Staccioli MP, Giordani P, Catalano V, et al. (2001). Inhibition of vascular endothelial growth factor by octreotide in colorectal cancer patients. Cancer Invest. 19 (1): 8–12.
- Chen M, Huang X, Yuen DS, Cheng CH (2011). A study on the functional interaction between the GH/PRL family of polypeptides with their receptors in zebrafish: Evidence against GHR1 being the receptor for somatolactin. Mol Cell Endocrinol. 337 (1–2): 114–121.
- Chiesa J, Ferrer C, Arnould C, Vouyovitch CM, Diaz JJ, Gonzalez S, et al. (2011). Autocrine Proliferative Effects of hGH Are Maintained in Primary Cultures of Human Mammary Carcinoma Cells. J Clin Endocrinol Metab. 96 (9): E1418–E1426.
- Cos S, Sánchez-Barceló EJ (2000). Melatonin and mammary pathological growth. Front Neuroendocrinol. 21 (2): 133–170.
- Cucina A, Proietti S, D’Anselmi F, Coluccia P, Dinicola S, Frati L, Bizzarri M (2009). Evidence for a biphasic apoptotic pathway induced by melatonin in MCF-7 breast cancer cells. J Pineal Res. 46 (2): 172–180.
- De Souza I, Morgan L, Lewis UL, Raggatt PR, Salih H, Hobbs JR (1974). Growth-hormone dependence among human breast cancers. Lancet. 2 (7874): 182–184.
- Di Bella G, Colori B (2009). Complete objective response of neuroblastoma to biological treatment. Neuro Endocrinol Lett. 30 (4): 437–449.
- Di Bella G, Madarena M (2009). Complete objective response of oesophageal squamocellular carcinoma to biological treatment. Neuro Endocrinol Lett. 30 (3): 312–321.
- Di Bella G (2008). Complete objective response to biological therapy of plurifocal breast carcinoma. Neuro Endocrinol Lett. 29 (6): 857–866.
- Di Bella G (2010). The Di Bella Method (DBM). Neuro Endocrinol Lett.. 31 (1): 1–42.
- Di Bella G (2005). “ Il Metodo Di Bella “ Mattioli Editore 3° Edizione.
- Di Bella L et al. (1994). Effect of Melatonin on circadian water intake by normal and tumor-bearing rats, Riunione cong. SIBS-SIF-SINU, Ischia.
- Di Bella L et al. (1980). Melatonin: an essential factor for the treatment and recovery from leukemia and cancer. International Symposium on Melatonin. N. Birau & W. Schloot. 161–162.
- Di Bella L (1988). Melatonin in cancer therapy. A satellite symposium of the 8th International Congress of Endocrinology, Hong Kong, Abstract from Symposium on melatonin and the pineal gland.
- Di Bella L (1974). Orientamenti fisiologici nella terapia delle emopatie.Boll. Sc. Med. 1–3.
- Di Bella L (1976). Physiological Basis for a rational therapy of bone marrow diseases. XVIth Internat. Congr. of Hematol. ,Kyoto. 9–45.
- Di Bella L, Gualano L (2006). Key aspects of melatonin physiology: thirty years of research. Neuro Endocrinol Lett. 27 (4): 425–432.
- Di Bella L (1997). Melatonina dalla ricerca agli interventi atti del Convegno Reggio Calabria– Atti del convegno – Reggio Calabria.
- Di Bella L, Rossi MT, Scalera G (1979). Perspectives in pineal functions. Prog Brain Res. 52 : 475–478.
- Di Bella L (1998). “Cancro: siamo sulla strada giusta?” Travel factory.
- Diakatou E, Kaltsas G, Tzivras M, Kanakis G, Papaliodi E, Kontogeorgos G (2011). Somatostatin and dopamine receptor profile of gastroenteropancreatic neuroendocrine tumors: an immuno-histochemical study. Endocr Pathol. 22 (1): 24–30.
- Dong C, Yuan L, Dai J, Lai L, Mao L, Xiang S, Rowan B, Hill SM (2010). Melatonin inhibits mitogenic cross-talk between retinoic acid-related orphan receptor alpha (RORalpha) and ERalpha in MCF-7 human breast cancer cells. Steroids. 75 (12): 944–951.
- Ferone D (2010). Somatostatin and dopamine receptors. Tumori. 96 (5): 802–805.
- Florio T, Morini M, Villa V, Arena S, Corsaro A, Thellung S, et al. (2003). Somatostatin inhibits tumor angiogenesis and growth via somatostatin receptor-3-mediated regulation of endothelial nitric oxide synthase and mitogen-activated protein kinase activities. Endocrinology. 144 (4): 1574–1584.
- Friend KE (2000). Targeting the growth hormone axis as a therapeuticstrategy in oncology. Growth Horm IGF Res. 10 A: S45–S46.
- Fürstenberger G, Morant R, Senn HJ (2003). Insulin-like growth factors and breast cancer. Onkologie. 26 (3): 290–294.
- Girgert R, Hanf V, Emons G, Gründker C (2010). Signal transduction of the melatonin receptor MT1 is disrupted in breast cancer cells by electromagnetic fields. Bioelectromagnetics. 31 (3): 237–245.
- Gruszka A, Pawlikowski M, Kunert-Radek J (2001). Anti-tumoral action of octreotide and bromocriptine on the experimental rat prolactinoma: anti-proliferative and pro-apoptotic effects. Neuro Endocrinol Lett. 22 (5): 343–348.
- Hagemeister AL, Sheridan MA (2008). Somatostatin inhibits hepatic growth hormone receptor and insulin-like growth factor I mRNA expression by activating the ERK and PI3K signalling pathways. Am J Physiol Regul Integr Comp Physiol. 295 (2) R490
- Hassan HT, Rees J (1990). Triple combination of retinoic acid plus actinomycin D plus dimethylformamide induces differentiation of human acute myeloid leukaemic blasts in primary culture. Cancer Chemother Pharmacol. 26(1): 26–30.
- Head KA (1998). Ascorbic acid in the prevention and treatment of cancer. Altern Med Rev. 3(3): 174–186.
- Held-Feindt J, Krisch B, Mentlein R (1999). Molecular analysis of the somatostatin receptor subtype 2 in human glioma cells. Brain Res Mol Brain Res. 64(1): 101–107.
- Hewitt SC, Li Y, Li L, Korach KS (2010). Estrogen-mediated regulation of Igf1 transcription and uterine growth involves direct binding of estrogen receptor alpha to estrogen-responsive elements. J Biol Chem. 285(4): 2676–2685.
- Hill SM (2011). Declining melatonin levels and MT1 receptor expression in aging rats is associated with enhanced mammary tumor growth and decreased sensitivity to melatonin. Breast Cancer Res Treat. 127(1): 91–98.
- Hill SM (2009). Molecular mechanisms of melatonin anticancer effects. Integr Cancer Ther. 8(4): 337–346.
- Hooghe R, Merchav S, Gaidano G, Naessens F, Matera L (1998). A role for growth hormone and prolactin in leukaemia and lymphoma? Cell Mol Life Sci. 54(10): 1095–1101.
- Hsieh TC, Elangovan S, Wu JM (2010). Differential suppression of proliferation in MCF-7 and MDA-MB-231 breast cancer cells exposed to alpha-, gamma- and delta-tocotrienols is accompanied by altered expression of oxidative stress modulatory enzymes. Anticancer Res. 30(10): 4169–4176.
- Israel K, Yu W, Sanders BG, Kline K (2000). Vitamin E succinate induces apoptosis in human prostate cancer cells: role for Fas in vitamin E succinate-triggered apoptosis. Nutr Cancer. 36(1): 90–100.
- Israel L (1996). Cancer as a survival program of individual cells inherited from prokaryotes, conserved but repressed in cells from higher organisms and unveiled by e nvironmental aggressions. Ann Med Interne (Paris). 147(6): 387–388.
- Jensen SS, Madsen MW, Lukas J, Binderup L, Bartek J (2001). Inhibitory effects of 1alpha,25-dihydroxyvitamin D(3) on the G(1)-S phase-controlling machinery. Mol Endocrinol. 15(8): 1370–1380.
- Jia WD, Xu GL, Xu RN, Sun HC, Wang L, Yu JH, et al. (2003). Octreotide acts as an antitumor angiogenesis compound and suppresses tumor growth in nude mice bearing human hepatocellular carcinoma xenografts. J Cancer Res Clin Oncol. 129(6): 327–334.
- Juul A (2001). The effects of oestrogens on linear bone growth. Hum Reprod Update. 7(3): 303–313.
- Kath R, Höffken K (2000). The significance of somatostatin analogues in the antiproliferative treatment of carcinomas. Recent Results Cancer Res. 153: 23–43.
- Kini AR, Peterson LA, Tallman MS, Lingen MW (2001). Angiogenesis in acute promyelocytic leukemia: induction by vascular endothelial growth factor and inhibition by all-trans retinoic acid. Blood. 97(12): 3919–3924.
- Kisker O, Onizuka S, Becker CM, Fannon M, Flynn E, D’Amato R, et al. (2003). Vitamin D binding protein-macrophage activating factor (DBP-maf) inhibits angiogenesis and tumor growth in mice. Neoplasia. 5(1): 32–40.
- Kleinberg DL, Ameri P, Singh B (2011). Pasireotide, an IGF-I action inhibitor, prevents growth hormone and estradiol-induced mammary hyperplasia. Pituitary. 14(1): 44–52.
- Klijn JG, Setyono-Han B, Bontenbal M, Seynaeve C, Foekens J (1996). Novel endocrine therapies in breast cancer. Acta Oncol. 35(5): 30–37.
- Korkmaz A, Sanchez-Barcelo EJ, Tan DX, Reiter RJ (2009). Role of melatonin in the epigenetic regulation of breast cancer. Res Treat. 115(1): 13–27.
- Kvetnoĭ IM, Levin IM (1986). Melatonin and tumor growth. Eksp Onkol. 8(4): 11–15.
- Lambert G, Estévez-Salmeron L, Oh S, Liao D, Emerson BM, Tlsty TD,et al. (2011). An analogy between the evolution of drug resistance in bacterial communities and malignant tissues. Nat Rev Cancer. 11(5): 375–382.
- Leung KC, Johannsson G, Leong GM, Ho KK (2004). Estrogen regulation of growth hormone action. Endocr Rev. 25(5): 693–721.
- Lin ZY (2010). Pharmacologic concentrations of melatonin have diverse influence on differential expressions of angiogenic chemokine genes in different hepatocellular carcinoma cell lines. Biomed Pharmacother. 64(10): 659–662.
- Lincoln DT, Sinowatz F, Temmim-Baker L, Baker HI, Kölle S, Waters MJ (1998). Growth hormone receptor expression in the nucleus and cytoplasm of normal and neoplastic cells. Histochem Cell Biol. 109(2): 141–159.
- Lissoni P, Eur J (1987). The clinical significance of melatonin serum determination in oncological patients and its correlations with GH and PRL blood levels. Cancer Clin Oncol. 23(7): 949– 957.
- Lissoni P, Rovelli F, Malugani F, Bucovec R, Conti A, Maestroni GJ (2001). Anti-angiogenic activity of melatonin in advanced cancer patients. Neuro Endocrinol Lett. 22(1): 45–47.
- Liu Y, Yang H, Otaka K, Takatsuki H, Sakanishi A (2005). Effects of vascular endothelial growth factor (VEGF) and chondroitin sulfate A on human monocytic THP-1 cell migration. Colloids Surf B Biointerfaces. 43(3–4): 216–220.
- Maestroni GJ, Hertens E, Galli P, Conti A, Pedrinis E (1996). Melatonin-induced T-helper cell hematopoietic cytokines resembling both interleukin-4 and dynorphin. J Pineal Res. 21(3): 131–139.
- Majewski S, Szmurlo A, Marczak M, Jablonska S, Bollag W (1994). Synergistic effect of retinoids and interferon alpha on tumor-induced angiogenesis: anti-angiogenic effect on HPV-harboring tumor-cell lines. Int J Cancer. 57(1): 81–85.
- Malafa MP, Fokum FD, Smith L, Louis A (2002). Inhibition of angiogenesis and promotion of melanoma dormancy by vitamin E succinate. Ann Surg Oncol. 9(10): 1023–1032.
- Manni A, Boucher AE, Demers LM, Harvey HA, Lipton A, Simmonds MA, et al. (1989). Endocrine effects of combined somatostatin analog and bromocriptine therapy in women with advanced breast cancer. Breast Cancer Res Treat. 14(3): 289–298.
- Mantell DJ, Owens PE, Bundred NJ, Mawer EB, Canfield AE (2000). 1 alpha,25-dihydroxyvitamin D(3) inhibits angiogenesis in vitro and in vivo. Circ Res. 87(3): 214–220.
- Mao L, Cheng Q, Guardiola-Lemaître B, Schuster-Klein C, Dong C, et al. (2010). In vitro and in vivo antitumor activity of melatonin receptor agonists. J Pineal Res. 49(3): 210–221.
- Mao L, Yuan L, Slakey LM, Jones FE, Burow ME, Hill SM.(2010). Inhibition of breast cancer cell invasion by melatonin is mediated through regulation of the p38 mitogen-activated protein kinase signaling pathway. Breast Cancer Res. 12(6): R107.
- Martínez-Campa C, González A, Mediavilla MD, Alonso-González C, Alvarez-García V, Sánchez-Barceló EJ, Cos S (2009). Melatonin inhibits aromatase promoter expression by regulating cyclooxygenases expression and activity in breast cancer cells. Br J Cancer. 101(9): 1613–1619.
- Mauri R, Lissoni P, Resentini M, De Medici C, Morabito F, Djemal S, et al. (1985). Effects of melatonin on PRL secretion during different photoperiods of the day in prepubertal and pubertal healthy subjects. J Endocrinol Invest. 8(4): 337–341.
- McMillan K, Perepelitsyn I, Wang Z, Shapshay SM (1999). Tumor growth inhibition and regression induced by photothermal vascular targeting and angiogenesis inhibitor retinoic acid. Cancer Lett. 137(1): 35–44.
- Mediavilla MD, Cos S, Sánchez-Barceló EJ (1999). Melatonin increases p53 and p21WAF1 expression in MCF-7 human breast cancer cells in vitro. Life Sci. 65(4): 415–420.
- Mediavilla MD, Sanchez-Barcelo EJ, Tan DX, Manchester L, Reiter RJ (2010). Basic mechanisms involved in the anti-cancer effects of melatonin. Med Chem. 17(36): 4462–4481.
- Mendoza RA, Enriquez MI, Mejia SM, Moody EE, Thordarson G (2010). Interactions between IGF-I, estrogen receptor-α (ERα), and ERβ in regulating growth/apoptosis of MCF-7 human breast cancer cells. J Endocrinol. 208(1): 1–9.
- Mentlein R, Eichler O, Forstreuter F, Held-Feindt J (2001). Somatostatin inhibits the production of vascular endothelial growth factor in human glioma cells. Int J Cancer. 92(4): 545–550.
- Mishima M, Yano T, Jimbo H, Yano N, Morita Y, Yoshikawa H, et al. (1999). Inhibition of human endometrial cancer cell growth in vitro and in vivo by somatostatin analog RC-160. Am J Obstet Gynecol. 181(3): 583–590.
- Murata A, Morishige F, Yamaguchi H (1982). Prolongation of survival times of terminal cancer patients by administration of large doses of ascorbate. Int J Vitam Nutr Res Suppl. 23: 103–113.
- Murray RD, Kim K, Ren SG (2004). Central and peripheral actions of somatostatin on the growth hormone-IGF-I axis. J Clin Invest. 114(3): 349–356.
- Neuzil J, Kagedal K, Andera L, Weber C, Brunk UT (2002). Vitamin E analogs: a new class of multiple action agents with anti-neoplastic and anti-atherogenic activity. Apoptosis. 7(2): 179–187.
- Onogi N, Okuno M, Matsushima-Nishiwaki R, Fukutomi Y, Moriwaki H, Muto Y, et al. (1998). Antiproliferative effect of carotenoids on human colon cancer cells without conversion to retinoic acid. Nutr Cancer. 32(1): 20–24.
- Ozerdem U, Stallcup WB (2004). Pathological angiogenesis is reduced by targeting pericytes via the NG2 proteoglycan. Angiogenesis. 7(3): 269–276.
- Piedrafita FJ, Pfahl M (1997). Retinoid-induced apoptosis and Sp1 cleavage occur independently of transcription and require caspase activation. Mol Cell Biol. 17(11): 6348–6358.
- Pinzani P, Orlando C, Raggi CC, Distante V, Valanzano R, Tricarico C, et al. (2001). Type-2 somatostatin receptor mRNA levels in breast and colon cancer determined by a quantitative RT-PCR assay based on dual label fluorogenic probe and the TaqMan technology. Regul Pept. 99(2–3): 79–86.
- Pollak M (1997). The potential role of somatostatin analogues in breast cancer treatment. Yale J Biol Med. 70(5–6): 535–539.
- Pumphrey CY, Theus AM, Li S, Parrish RS, Sanderson RD (2002). Neoglycans, carbodiimide-modified glycosaminoglycans: a new class of anticancer agents that inhibit cancer cell proliferation and induce apoptosis. Cancer Res. 62(13): 3722–3728.
- Radman M (1975). SOS repair hypothesis: phenomenology of an inducible DNA repair which is accompanied by mutagenesis. Basic Life Sci. 5A: 355–367.
- Rögelsperger O, Ekmekcioglu C, Jäger W, Klimpfinger M, Königsberg R, Krenbek D, et al. (2009). Coexpression of the melatonin receptor 1 and nestin in human breast cancer specimens. J Pineal Res. 46(4): 422–432.
- Sall JW, Klisovic DD, O’Dorisio MS (2004). Somatostatin inhibits IGF-1 mediated induction of VEGF in human retinal pigment epithelial cells. Exp Eye Res. 79(4): 465–476.
- Sandret L, Maison P, Chanson P (2011). Place of Cabergoline in Acromegaly: A Meta-Analysis. J Clin Endocrinol Metab. 96(5):1327–1335.
- Saveanu A, Muresan M, De Micco C, Taieb D, Germanetti AL, Sebag F, Henry JF, Brunaud L, Enjalbert A, Weryha G, Barlier A (2011). Expression of Somatostatin receptors, dopamine D2 receptors, noradrenaline transporters, and vesicular monoamine transporters in 52 pheochromocytomas and paragangliomas. Endocr Relat Cancer. 18(2): 287–300.
- Schaer JC, Waser B, Mengod G, Reubi JC (1997). Somatostatin receptor subtypes sst1, sst2, sst3 and sst5 expression in human pituitary, gastroentero-pancreatic and mammary tumors: comparison of mRNA analysis with receptor autoradiography. Int J Cancer. 70(5): 530–537.
- Schally AV, Comaru-Schally AM, Nagy A, Kovacs M, Szepeshazi K, Plonowski A, et al. (2001). Hypothalamic hormones and cancer. Front Neuroendocrinol. 22(4): 248–291.
- Schally AV, Nagy A (2003). New approaches to treatment of various cancers based on cytotoxic analogs of LHRH, somatostatin and bombesin. Life Sci. 72(21): 2305–2320.
- Shklar G, Schwartz JL (1996). Vitamin E inhibits experimental carcinogenesis and tumour angiogenesis. Eur J Cancer B Oral Oncol. 32B(2): 114–119.
- Szepesházi K, Halmos G, Schally AV, Arencibia JM, Groot K, Vadillo-Buenfil M, et al. (1999). Growth inhibition of experimental pancreatic cancers and sustained reduction in epidermal growth factor receptors during therapy with hormonal peptide analogs. J Cancer Res Clin Oncol. 125(8–9): 444–452.
- Taboada GF, Neto LV, Luque RM, Córdoba-Chacón J, de Oliveira Machado E, de Carvalho DP, et al. (2011). Impact of gsp oncogene on the mRNA content for somatostatin and dopamine receptors in human somatotropinomas. Neuroendocrinology. 93(1): 40–47.
- Tang FY, Meydani M (2001). Green tea catechins and vitamin E inhibit angiogenesis of human microvascular endothelial cells through suppression of IL-8 production. Nutr Cancer. 41(1–2): 119–125.
- Taslipinar A, Bolu E, Kebapcilar L (2001). Insulin-like growth factor-1 is essential to the increased mortality caused by excessgrowth hormone: a case of thyroid cancer and non-Hodgkin’slymphoma in a patient with pituitary acromegaly. Med Oncol. 26(1): 62–66.
- Turley JM, Funakoshi S, Ruscetti FW, Kasper J, Murphy WJ, Longo DL, et al. (1995). Growth inhibition and apoptosis of RL human B lymphoma cells by vitamin E succinate and retinoic acid: role for transforming growth factor beta. Cell Growth Differ. 6(6): 655–663.
- Turner HE, Nagy Z, Gatter KC, Esiri MM, Harris AL, Wass JA (2000).Angiogenesis in pituitary adenomas – relationship to endocrine function, treatment and outcome. J Endocrinol. 165(2): 475–481.
- Uray IP, Brown PH (2011). Chemoprevention of hormone receptor-negative breast cancer: new approaches needed. Recent Results Cancer Res. 188: 147–162.
- van Eijck CH, Kwekkeboom DJ, Krenning EP (1998). Somatostatin receptors and breast cancer. Q J Nucl Med. 42(1): 18–25.
- Vidal S, Oliveira MC, Kovacs K, Scheithauer BW, Lloyd R (2000). Immunolocalization of vascular endothelial growth factor in the GH3 cell line. Cell Tissue Res. 300(1): 83–88.
- Voigt A, Hartmann P, Zintl F (2000). Differentiation, proliferation and adhesion of human neuroblastoma cells after treatment with retinoic acid. Cell Adhes Commun. 7(5): 423–440.
- Watson JC, Balster DA, Gebhardt BM, O’Dorisio TM, O’Dorisio MS, Espenan GD, et al. (2001). Growing vascular endothelial cells express somatostatin subtype 2 receptors. Br J Cancer. 85(2): 266–272.
- Wennbo H, Törnell J (2000). The role of prolactin and growth hormone in breast cancer. Oncogene. 19(8): 1072–1076.
- Wiedermann CJ, Reinisch N, Braunsteiner H (1993). Stimulation of monocyte chemotaxis by human growth hormone and its deactivation by somatostatin. Blood. 82(3): 954–960.
- Xu J, Zhang Y, Berry PA, Jiang J, Lobie PE, Langenheim JF, Chen WY, Frank SJ (2011). Growth hormone signaling in human T47D breast cancer cells: potential role for a growth hormone receptor-prolactin receptor complex. Mol Endocrinol. 25(4): 597–610.
- Yarman S, Kurtulmus N, Canbolat A, Bayindir C, Bilgic B, Ince N (2010). Expression of Ki-67, p53 and vascular endothelial growth factor (VEGF) concomitantly in growth hormone-secreting pituitary adenomas; which one has a role in tumor behavior? NeuroEndocrinol Lett. 31(6): 823–828.
- Zeitler P Siriwardana G (2000). Stimulation of mitogen-activated protein kinase pathway in rat somatotrophs by growth hormone-releasing hormone. Endocrine. 12(3): 257–264.
- Zhou DW, Li SW, Wang XH, Zheng XM, Ding XG (2011). Estradiol stimulates proliferation of prostatic smooth muscle cells via estrogen receptor alpha and IGF1. Zhonghua Nan Ke Xue. 17(2): 131–135.