Studio retrospettivo sul Carcinoma Mammario trattato unicamente con MDB (terapia di prima-linea)
Pubblicato il 23/09/2014
Studio clinico rettrospettivo sulla sicurezza ed efficacia del trattamento di prima linea con Somatostatina abbinata a Retinoidi, Melatonina, Vit. D3, e basse dosi di Ciclofosfamide, in venti casi di Carcinoma Mammario.
Giuseppe Di Bella, Fabrizio Mascia, Alessandro Ricchi, Biagio Colori*
Di Bella Foundation, Via Guglielmo Marconi 51, 40122, Bologna, IT.-
Rizzoli Institute, Scientific Research and Care Institute, Bologna, IT.
Di Bella Foundation, Via Guglielmo Marconi 51, 40122, Bologna, IT.-
Rizzoli Institute, Scientific Research and Care Institute, Bologna, IT.
INTRODUZIONE
Nei paesi occidentali il tumore della mammella costituisce la più frequente neoplasia femminile: nel solo periodo 1998-2002 ha rappresentato il 24,9% del totale delle diagnosi di tumore. Anche in termini di mortalità è risultata la prima fra le cause tumorali, con il 17,1% del totale dei decessi.
Nell'anno 2012 è stato stimato che delle 226.870 donne alle quali è stata data una diagnosi di tumore, 39.510 moriranno di cancro al seno. Le stime dell' Italia mostrano come, a un anno dalla diagnosi, il tasso di sopravvivenza relativa è del 95% per gli uomini e del 97% per le donne. Questo valore decresce col passare del tempo e a distanza di 5 anni la probabilità di sopravvivere a un tumore alla mammella è dell'85% per entrambi i sessi.
Quasi tutte le donne con un tumore del seno, indipendentemente dallo stadio, subiscono, se le condizioni cliniche lo consentono, un intervento chirurgico per la rimozione della lesione. Nei casi in cui ciò sia possibile si ricorre alla chirurgia conservativa (quadrantectomia) sovente seguita da radioterapia
adiuvante a scopo preventivo. Durante l'intervento, si può altresì procedere all'asportazione di potenziali linfonodi maligni in sede ascellare ("tecnica del linfonodo sentinella").
Nella maggior parte delle pazienti con carcinoma in situ, così come negli stadi precoci (stadio I-II-IIIA), risulta talvolta necessario asportare più di un quadrante di seno mediante mastectomia parziale o segmentale: anch'essa seguita da terapia radiante. Le forme più avanzate (carcinoma metastatico, stadi IIIB-IIIC-IV), vengono trattate con l'asportazione dell'intero seno (mastectomia radicale modificata).
Ad oggi, l'asportazione chirurgica dei tessuti tumorali è l'unico mezzo ufficialmente riconosciuto per raggiungere, negli stadi non avanzati, la guarigione. Tuttavia, anche nei casi circoscritti, l'intervento chirurgico può esporre al rischio di distacco, dalle sezioni marginali, di residue cellule tumorali in grado di diffondersi in altri distretti corporei attraverso il flusso ematico, determinando successive disseminazioni a lungo raggio. Per tali ragioni, la resezione chirurgica della lesione tumorale viene associata con chemioterapia adiuvante e neo-adiuvante.
Le altre strategie terapeutiche prevedono l'impiego di molecole con blocco specifico del rilascio degli ormoni sessuali (tamoxifene). La prassi clinica prevede che questi vengano impiegati a seguito della asportazione della lesione e della successiva indagine immuno-istochimica per valutazione della presenza di recettori per gli estrogeni (ER) e progesterone (PR). In caso di positività vengono suggerite specifiche molecole.
Sebbene questo farmaco riduca drasticamente le ricadute, è ormai assodato come sia in grado di indurre, di fatto, una menopausa su base chimica, nonché pericolosi fenomeni tromboembolitici, diminuzione dell'attività cognitiva ed un aumento del rischio di sviluppo di neoplasie (carcinoma dell’endometrio): tale rischio quadruplica dopo i canonici cinque anni di trattamento.
In sostituzione al tamoxifene, vengono impiegate molecole con medesima funzione (inibitori delle aromatasi: anastrozolo, letrozolo), ma con differente meccanismo d'azione. Attualmente, quest'ultima classe di farmaci è riservata esclusivamente alle donne in età post-menopausale. Un ulteriore contributo, grazie agli avanzamenti delle conoscenze e delle tecniche di biologia molecolare, viene fornito gli anticorpi monoclonali. Un esempio è il trastuzumab, anticorpo monoclonale selettivo per i recettori del fattore di crescita epidermico (EGFR).
Sebbene i suddetti trattamenti abbiano favorito nel corso del tempo maggiori miglioramenti in termini di sopravvivenza, rimane tutt'ora discutibile il loro approccio, sovente invasivo, demolitivo, e a scapito delle condizioni psicosomatiche generali delle pazienti.
Prima di tutto, la chirurgia rimane di fatto un intervento mutilante e traumatico, anche in caso di trattamento conservativo, mentre il contributo dei regimi polichemioterapici si riduce a minime percentuali. Negli ultimi si sta valutando l'impiego giornaliero di basse dosi di chemioterapici per os (chemioterapia metronomica), con lo scopo sia di esaltare l'azione del farmaco sia di evitare gli effetti collaterali.
Dato ancor più interessante, negli ultimi decenni numerose molecole biologiche, tra cui la somatostatina e analoghi, la melatonina e differenti classi di vitamine, quali Retinoidi e Vitamina D3 e Tocoferoli, si sono mostrate potenzialmente utili nel trattamento preventivo e terapeutico del tumore al seno (Seitz et al. 2013; Sanchez-
Barcelo et al. 2012; Tang & Gudas 2012; Mehta et al. 2012; Frati et al. 2011; Fulan et al. 2011;). Numerosi studi in vitro condotti su svariate linee cellulari, ne hanno evidenziato le spiccate attività antitumorali, chiarendo da una parte i meccanismi d'azione, e aprendo la strada verso il raggiungimento di risultati incoraggianti nella pratica clinica (Proietti et al. 2012; Margheri et al. 2012; Cescon et al. 2012; Ostendorf et al. 2012; Suhail et al. 2012; Zhang et al. 2012; Proietti at al. 2011; He et al. 2009; Watt et al. 2009; Lee et al 2008). Tuttavia, sono limitati gli studi clinici a lungo termine sull'uomo e sulla loro associazione in un contesto multiterapico che ne esalti il sinergismo e l'interattività .
Gli autori riportano di seguito un studio clinico osservazionale retrospettivo condotto su 20 pazienti affette da carcinoma mammario, alle quali è stata somministrata la terapia biologia (Metodo Di Bella, DBM), mediante l'uso combinato a basse dosi di ciclofosfamide ed inibitori estrogenici più molecole ad appurata azione anti-tumorale, quali somatostatina/octreotide, melatonina, inibitori prolattinici, Retinoidi, Vitamine, E,C, D3, Calcio e componenti della matrice extracellulare (Di Bella 2001).
Nell'anno 2012 è stato stimato che delle 226.870 donne alle quali è stata data una diagnosi di tumore, 39.510 moriranno di cancro al seno. Le stime dell' Italia mostrano come, a un anno dalla diagnosi, il tasso di sopravvivenza relativa è del 95% per gli uomini e del 97% per le donne. Questo valore decresce col passare del tempo e a distanza di 5 anni la probabilità di sopravvivere a un tumore alla mammella è dell'85% per entrambi i sessi.
Quasi tutte le donne con un tumore del seno, indipendentemente dallo stadio, subiscono, se le condizioni cliniche lo consentono, un intervento chirurgico per la rimozione della lesione. Nei casi in cui ciò sia possibile si ricorre alla chirurgia conservativa (quadrantectomia) sovente seguita da radioterapia
adiuvante a scopo preventivo. Durante l'intervento, si può altresì procedere all'asportazione di potenziali linfonodi maligni in sede ascellare ("tecnica del linfonodo sentinella").
Nella maggior parte delle pazienti con carcinoma in situ, così come negli stadi precoci (stadio I-II-IIIA), risulta talvolta necessario asportare più di un quadrante di seno mediante mastectomia parziale o segmentale: anch'essa seguita da terapia radiante. Le forme più avanzate (carcinoma metastatico, stadi IIIB-IIIC-IV), vengono trattate con l'asportazione dell'intero seno (mastectomia radicale modificata).
Ad oggi, l'asportazione chirurgica dei tessuti tumorali è l'unico mezzo ufficialmente riconosciuto per raggiungere, negli stadi non avanzati, la guarigione. Tuttavia, anche nei casi circoscritti, l'intervento chirurgico può esporre al rischio di distacco, dalle sezioni marginali, di residue cellule tumorali in grado di diffondersi in altri distretti corporei attraverso il flusso ematico, determinando successive disseminazioni a lungo raggio. Per tali ragioni, la resezione chirurgica della lesione tumorale viene associata con chemioterapia adiuvante e neo-adiuvante.
Le altre strategie terapeutiche prevedono l'impiego di molecole con blocco specifico del rilascio degli ormoni sessuali (tamoxifene). La prassi clinica prevede che questi vengano impiegati a seguito della asportazione della lesione e della successiva indagine immuno-istochimica per valutazione della presenza di recettori per gli estrogeni (ER) e progesterone (PR). In caso di positività vengono suggerite specifiche molecole.
Sebbene questo farmaco riduca drasticamente le ricadute, è ormai assodato come sia in grado di indurre, di fatto, una menopausa su base chimica, nonché pericolosi fenomeni tromboembolitici, diminuzione dell'attività cognitiva ed un aumento del rischio di sviluppo di neoplasie (carcinoma dell’endometrio): tale rischio quadruplica dopo i canonici cinque anni di trattamento.
In sostituzione al tamoxifene, vengono impiegate molecole con medesima funzione (inibitori delle aromatasi: anastrozolo, letrozolo), ma con differente meccanismo d'azione. Attualmente, quest'ultima classe di farmaci è riservata esclusivamente alle donne in età post-menopausale. Un ulteriore contributo, grazie agli avanzamenti delle conoscenze e delle tecniche di biologia molecolare, viene fornito gli anticorpi monoclonali. Un esempio è il trastuzumab, anticorpo monoclonale selettivo per i recettori del fattore di crescita epidermico (EGFR).
Sebbene i suddetti trattamenti abbiano favorito nel corso del tempo maggiori miglioramenti in termini di sopravvivenza, rimane tutt'ora discutibile il loro approccio, sovente invasivo, demolitivo, e a scapito delle condizioni psicosomatiche generali delle pazienti.
Prima di tutto, la chirurgia rimane di fatto un intervento mutilante e traumatico, anche in caso di trattamento conservativo, mentre il contributo dei regimi polichemioterapici si riduce a minime percentuali. Negli ultimi si sta valutando l'impiego giornaliero di basse dosi di chemioterapici per os (chemioterapia metronomica), con lo scopo sia di esaltare l'azione del farmaco sia di evitare gli effetti collaterali.
Dato ancor più interessante, negli ultimi decenni numerose molecole biologiche, tra cui la somatostatina e analoghi, la melatonina e differenti classi di vitamine, quali Retinoidi e Vitamina D3 e Tocoferoli, si sono mostrate potenzialmente utili nel trattamento preventivo e terapeutico del tumore al seno (Seitz et al. 2013; Sanchez-
Barcelo et al. 2012; Tang & Gudas 2012; Mehta et al. 2012; Frati et al. 2011; Fulan et al. 2011;). Numerosi studi in vitro condotti su svariate linee cellulari, ne hanno evidenziato le spiccate attività antitumorali, chiarendo da una parte i meccanismi d'azione, e aprendo la strada verso il raggiungimento di risultati incoraggianti nella pratica clinica (Proietti et al. 2012; Margheri et al. 2012; Cescon et al. 2012; Ostendorf et al. 2012; Suhail et al. 2012; Zhang et al. 2012; Proietti at al. 2011; He et al. 2009; Watt et al. 2009; Lee et al 2008). Tuttavia, sono limitati gli studi clinici a lungo termine sull'uomo e sulla loro associazione in un contesto multiterapico che ne esalti il sinergismo e l'interattività .
Gli autori riportano di seguito un studio clinico osservazionale retrospettivo condotto su 20 pazienti affette da carcinoma mammario, alle quali è stata somministrata la terapia biologia (Metodo Di Bella, DBM), mediante l'uso combinato a basse dosi di ciclofosfamide ed inibitori estrogenici più molecole ad appurata azione anti-tumorale, quali somatostatina/octreotide, melatonina, inibitori prolattinici, Retinoidi, Vitamine, E,C, D3, Calcio e componenti della matrice extracellulare (Di Bella 2001).
MATERIALI E METODI
Criteri di arruolamento.
Nella presente investigazione sono state arruolate esclusivamente le pazienti con un Eastern Cooperative Oncology Group (ECOG) status 2 , con diagnosi istologica di tumore al seno e con caratteristiche di malattia misurabile in accordo con i Response Evaluation Criteria in Solid Tumors (RECIST) (Patrick et al. 2000).
Altro requisito ai fini dell'arruolamento è stato l'assenza dei regimi terapeutici standard (interventi chirurgici, polichemioterapie, terapie radianti, anticorpi monoclonali), e di aver accettato, previo consenso informato, la somministrazione della terapia biologica di prima linea.
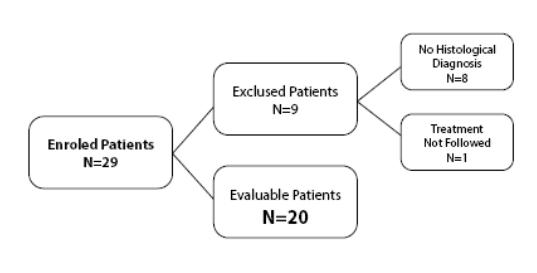
Il campione delle pazienti sopradescritto è stato ulteriormente suddiviso in due sottogruppi, in funzione dello stadio della patologia neoplastica:
Gruppo A: Pazienti in stadio precoce di neoplasia mammaria (stadi I, II, IIIA);
Gruppo B: Pazienti con malattia Localmente Avanzata/Metastatica (stadi IIIB, IIIC, IV).
Trattamento.
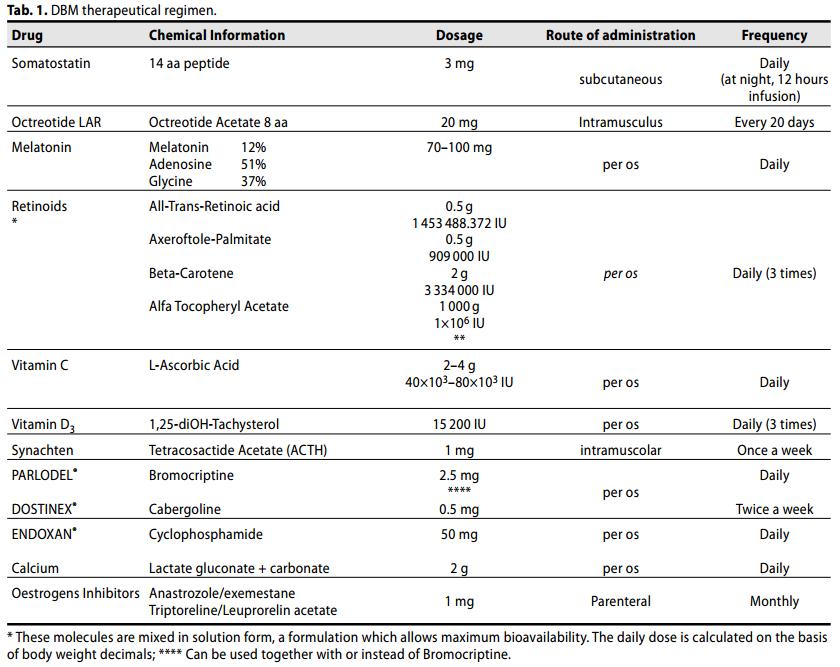
Tutte le pazienti hanno ricevuto una combinazione giornaliera di Somatostatina/octreotide, Melatonina, Retinoidi solubilizzati in Alfa Tocoferile Acetato, agonisti dopaminergici, inibitori estrogenici e dosaggi minimali di ciclofosfamide.
Nel dettaglio, sono state somministrate nelle seguenti modalità:
dosaggio graduale per os di:
Gruppo A: Pazienti in stadio precoce di neoplasia mammaria (stadi I, II, IIIA);
Gruppo B: Pazienti con malattia Localmente Avanzata/Metastatica (stadi IIIB, IIIC, IV).
Trattamento.
Tutte le pazienti hanno ricevuto una combinazione giornaliera di Somatostatina/octreotide, Melatonina, Retinoidi solubilizzati in Alfa Tocoferile Acetato, agonisti dopaminergici, inibitori estrogenici e dosaggi minimali di ciclofosfamide.
Nel dettaglio, sono state somministrate nelle seguenti modalità:
dosaggio graduale per os di:
- Ac. Retinoico (gr 0,25)(ATRA, 1 453 488.372 IU), + axeroftolo palmitato (gr 0,25)(909000 IU) + Betacarotene (gr. 1)(3 334 000 IU) solubilizzati in alfa tocoferile acetato (gr 500, rapporto stechiometrico 1:1:4:2)(1×106 IU); una volta al dì per sette giorni a digiuno, dopo altri 7 giorni due volte al giorno; dalla terza settimana, per l'intera durata del trattamento, 3 volte al giorno, per os;
- diidrotachisterolo (10 gocce)(15 200 IU) in concomitanza con i Retinoidi;
- Somatostatina (1mg) somministrazione scalare (1 mg nei primi 7 giorni, con somministrazione finale di 3 mg a 21 giorni di trattamento)
- tetracosactide esacetato (1 mg) somministrazione iniziale intramuscolare ogni 7 giorni, monitorando costantemente pressione e glicemia, dopo 30 giorni 1\2 fiala settimanale;
- Octreotide a lento rilascio LAR (20 mg) ogni 3 settimane intramuscolo;
- Melatonina da 5 mg per os: 10 mg i al mattino, a mezzodì, la sera ai pasti oltre a 40 mg prima di coricarsi (dosaggio giornaliero complessivo medio = 70 mg);
- Cabergolina per os durante il pasto principale 1 mg (uguale a 1\2 compressa) 2 volte la settimana;
- Bromocriptina (2,5 mg) per os mezza compressa mattino e sera;
- Ciclofosfamide (50-100 mg) per os, dosaggio graduale: iniziare con 1 compressa al pasto principale, dopo una settimana 1cpr mattino e sera;
- Acido Ascorbico (Vit C) per os, dosaggio graduale: 1/2 cucchiaino( (2 g = 4×103 IU)) in un bicchiere d'acqua mezzodì e sera durante il pasto insieme a
- Calcio lattato gluconato +calcio carbonato pari a 1000 mg di calcio nello stesso bicchiere;
- Condroitinsolfato (500 mg) una capsula mattino mezzodì e sera ai pasti;
Maggiori dettagli inerenti alle modalità di somministrazione e alle rispettive posologie sono illustrati nella tab.1:
Valutazione della risposta al trattamento delle lesioni target (Efficacia):
Metodi Statistici e Analitici
I criteri di valutazione alla risposta obiettiva fanno riferimento alle linee guida adottate dalla World Health Organization (WHO handbook) e dai tassi di risposta Response Evaluation Criteria In Solid Tumors (RECIST). Questi sono classificati in:
- Risposta Globale (OR);
- Risposta Completa (CR);
- Risposta Parziale (PR);
- Malattia Progressiva (P);
- Malattia Stabile (SD),
Nelle analisi di sopravvivenza, sono state valutate la sopravvivenza complessiva (Overall Survival, OS) e la sopravvivenza libera da progressione (Progression Free Survival, PFS). Queste ultime analisi sono state effettuate mediante il metodo Kaplan-Meier, con un C.I. del 95% ed analizzate con l'ausilio del pacchetto software (versione 2.15.12, 2012).
Valutazione della Sicurezza e della Tossicità:
Per la valutazione della tossicità sono stati considerati esclusivamente gli eventi avversi potenzialmente correlabili al trattamento (gradi di correlazione: possibile, probabile o certo, espressi come frequenza assoluta (n), relativa (%), ed intervallo di confidenza (CI) al 95%), come descritto dal National Cancer Institute (NCICTC) criteria (http://www.eortc.be/services/doc/ctc/).
Nota: questo è uno studio sull'uso combinato di farmaci che hanno già superato tutti i test di affidabilità e attività antitumorale. Pertanto, poiché tutti i farmaci sono già ampiamente testati e il cui uso è approvato dalle organizzazioni sanitarie internazionali, ma in questo contesto sono utilizzati solo in una nuova combinazione, è stato deciso di non sottoporsi a qualsiasi comitato etico.
Questo studio è stato condotto in accordo con le direttive stabilite dal The Good Clinical Practices directives (Sackett et al. 1996) e dalla Dichiarazione di Helsinki.
Tutte le pazienti hanno pertanto dato il proprio consenso informato per la partecipazione allo studio.
RISULTATI
Un totale di 29 pazienti sono state consecutivamente trattate con terapia biologica (MDB) e monitorate dal periodo Gennaio 2009 - Dicembre 2012. Di queste, venti (20) hanno soddisfatto i criteri di valutazione e sono state arruolate all'interno della indagine restrospettiva.
Per la valutazione della tossicità sono stati considerati esclusivamente gli eventi avversi potenzialmente correlabili al trattamento (gradi di correlazione: possibile, probabile o certo, espressi come frequenza assoluta (n), relativa (%), ed intervallo di confidenza (CI) al 95%), come descritto dal National Cancer Institute (NCICTC) criteria (http://www.eortc.be/services/doc/ctc/).
Nota: questo è uno studio sull'uso combinato di farmaci che hanno già superato tutti i test di affidabilità e attività antitumorale. Pertanto, poiché tutti i farmaci sono già ampiamente testati e il cui uso è approvato dalle organizzazioni sanitarie internazionali, ma in questo contesto sono utilizzati solo in una nuova combinazione, è stato deciso di non sottoporsi a qualsiasi comitato etico.
Questo studio è stato condotto in accordo con le direttive stabilite dal The Good Clinical Practices directives (Sackett et al. 1996) e dalla Dichiarazione di Helsinki.
Tutte le pazienti hanno pertanto dato il proprio consenso informato per la partecipazione allo studio.
RISULTATI
Un totale di 29 pazienti sono state consecutivamente trattate con terapia biologica (MDB) e monitorate dal periodo Gennaio 2009 - Dicembre 2012. Di queste, venti (20) hanno soddisfatto i criteri di valutazione e sono state arruolate all'interno della indagine restrospettiva.
L'età mediana era di 51 anni (intervallo: 30-70 anni), con un ECOG status 2. Il principale istotipo della lesione primaria è stato carcinoma duttale infiltrante (IDC, 15 casi, 75 %), mentre fegato, linfonodi e ossa costituivano le principali sedi di lesione secondaria (10 %, 60% e 30 % rispettivamente).
il profilo biologico molecolare è solo valutabile nel 60% dei pazienti (12 casi).
Di seguito gli stadi della malattia al momento dell'arruolamento:
il profilo biologico molecolare è solo valutabile nel 60% dei pazienti (12 casi).
Di seguito gli stadi della malattia al momento dell'arruolamento:
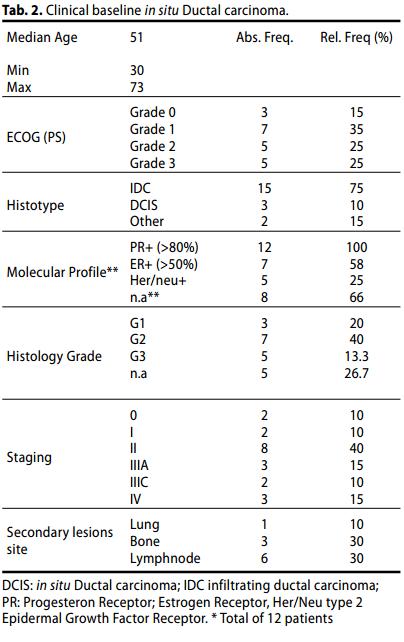
Malattia precoce (Gruppo A):
- Stadio 0, 2 casi (10%);
- Stadio I, 2 casi (10%);
- Stadio II, 8 casi (40%)
I Grading erano: G1 20%; G2 40%, G3 13,3 %, n.d. (26,7 %).
Malattia Localmente avanzata/Metastatica (Gruppo B):
- Stadio IIIA, 3 casi (15%);
- Stadio IIIC, 2 casi (10%);
- Stadio IV, 3 casi (15%).
- Di questi, 4 casi (80%) mostravano un grading G3.
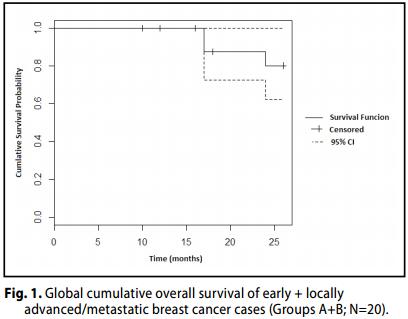
Complessivamente, la risposta globale obiettiva (OR) [Risposta completa (CR) + risposta parziale (PR)] è stata osservata nel 75% (15 casi; 53.1- 88.8; 95% CI) delle pazienti, con una CR nel 55% dei casi (n=11; 30-70; 95% CI)
Inoltre, l' 85.0% (17 casi; 63.9-94.7; 95% CI) delle pazienti ha ottenuto un beneficio clinico obiettivo. [CR+PR+SD].
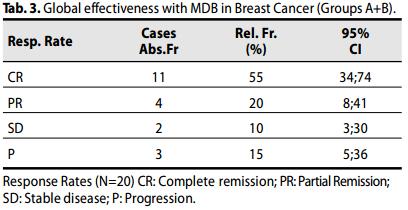
Il tasso di sopravvivenza globale (OS) delle pazienti è stato del 100% .
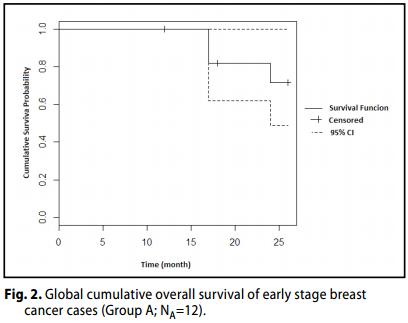
Group A (Carcinoma mammario in fase iniziale, I - II - IIIA Stadio).
Un OR (CR + PR) è stato raggiunto nel 91% dei pazienti (7 + 4 casi; 64-98; 95% CI), con CR nel 58% dei casi (n = 7, 32-81; 95% CI). .
In aggiunta, tutte le pazienti hanno ottenuto un beneficio clinico [CR+PR+SD]
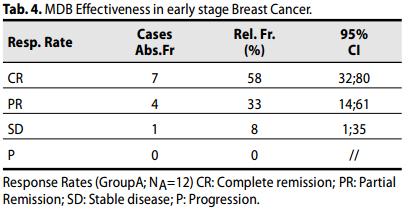
Il tasso di sopravvivenza globale (OS) delle pazienti è stato del 100%.
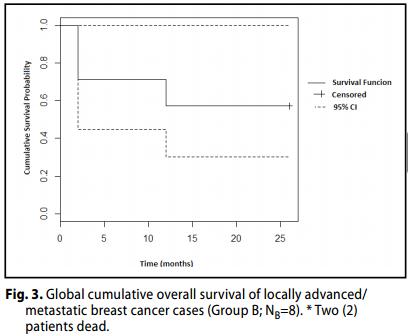
Group B (Carcinoma Localmente avanzato / metastatico, III B - III C- IV Stages).
La OR (CR + PR) è stato del 50% (3 + 1 dei casi, 21-78, 95% CI) dei pazienti, con CR a 37,5% dei casi. Inoltre, il 62,5% dei pazienti ha raggiunto un beneficio clinico (CR + PR + SD).
Tre (3) casi (60%) risultavano in progressione P (23-88; 95% CI).
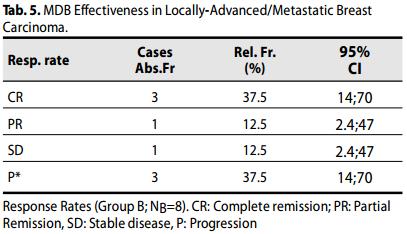
Il tasso di sopravvivenza globale (OS) delle pazienti è stato del 71%.
Valutazione della sicurezza
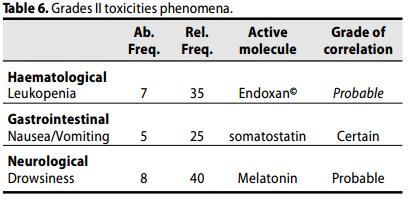
I più frequenti fenomeni di tossicità rilevati nello studio, di grado I e II, sono stati I seguenti:
tossicità ematologica: Leucopenia, 35%, gastrointestinale: nausea 25%,e sonnolenza (40%).
Questi fenomeni sono stati gli unici osservati.
Una riduzione, ritardo, o sospensione temporanea del trattamento a causa di tossicità si è resa necessaria nei pazienti leucopenia (Sospensione di Ciclofosfamide sino a ripristino della conta leucocitaria), e nei casi di effetti gastrointestinali
Non sono stati riscontrati decessi associati con il trattamento.
DISCUSSIONE
Razionale della Terapia e discussione dei dati clinici.
I fenomeni di proliferazione incontrollata e la perdita di differenziazione, anche se in misura diversa, sono denominatori comuni a tutte le neoplasie. Numerose investigazioni cliniche, nonché studi sperimentali, indicano chiaramente come alcuni ormoni pituitari, incluso il l'ormone della crescita (GH) e la Prolattina (PRL), abbiano un ruolo cruciale nello sviluppo e nella progressione del tumore al seno umano (Schally et al. 2001; Kamenicky et al. 2010; Perry et al., 2008; Harvey 2012;
Bernichtein et al. 2010; Faupel-Badger et al. 2010; Raccurt et al. 2002). Se da una parte l'espressione recettoriale ubiquitaria della Prolattina e del GH rappresenta una delle chiare conferme del ruolo mitogeno diretto e generalizzato di questi ormoni (Wu et al. 2011; Xu et al. 2012), d'altro canto è stato dimostrato il rapporto causale dose-dipendente, tra l'espressione recettoriale del GH e i processi di induzione e progressione tumorale. Infatti la concentrazioni di GHR e di GHRHR e' nettamente superiore nei tessuti tumorali del seno rispetto alla loro espressione recettoriale fisiologica e peritumorale, a ulteriore conferma del potente ruolo mitogeno del GH con un indice proliferativo dose-dipendente (Gebre-Medhin et al. 2001; Chatzistamou et al. 2004)..
Attualmente è in fase di studio la meccanica temporale di tale processo etiopatogenetico: tra i più verosimili, sono stati suggeriti probabili meccanismi di segnalazione autocrina e/o paracrina, sulla base della rilevazione della produzione locale del GH e del IGFI sui tessuti di Carcinoma mammario, peritumorale e fibroblasti. (Kaulsay et al. 2001; Siriwardana et al. 2010)]. Ne consegue che la proliferazione cellulare è strettamente dipendente dall'asse Prolattina - GH, e da molecole mitogene GH dipendenti, da esso positivamente regolate, come l'Epidermal Growth Factor (EGF), il Fibroblast Growth Factor (FGF), l'Hepatocyte Growth Facor (HGF), L'Insulin Growth Factor (IGF1-2), il Nerve Growth Factor (NGF), il Platelet Derived Growth Factor (PDGF), il Vascular Endothelial Growth Factor (VEGF), il Trasforming Growth Factor (TGF), oltre che da fattori di crescita prodotti dall'apparato gastrointestinale, come il Peptide Vasoattivo Intestinale (VIP), la Colecistochinina (CCK) (Li et al. 2011; Frank et al. 2008; Fürstenberger et al. 2003; Laban et al. 2003; Milewicz et al. 2011; Carver et al. 2010; Haines et al. 2009; Smith et al. 2011; Moody et al. 2003).
Sia la proliferazione cellulare fisiologica che neoplastica, avvengono per mezzo di queste stesse molecole, che la cellula neoplastica utilizza in misura moltiplicata rispetto a quella sana. Da ciò si deduce come l'asse PRL-GH-IGF1 abbia una determinate influenza sullo sviluppo biologico neoplastico. Appare pertanto evidente come l'utilizzo di antagonisti biologici del GH, come Somatostatina e analoghi, non riducano solo l'espressione e la trascrizione di fattori di crescita altamente mitogeni, come IGF1-2, EGF, FGF, VEGF, PDGF, ma estendono la loro regolazione negativa alle vie di segnalazione dei rispettivi bersagli recettoriali, con conseguenti riflessi antiproliferativi e antiangiogenici. Tale concezione sta lentamente emergendo attraverso le sempre più frequenti ricerche di base, sebbene ancora siano scarsi i modelli in vivo.
Le neoplasie mammarie rispondono mitogenicamente a IGF mediante la via di segnalazione innescata dal suo recettore IGFR. L'effetto soppressivo della SST e analoghi, sui livelli sierici di IGF1, è sia diretto, attraverso l'inibizione del gene di IGF, che indiretto, mediante la soppressione del GH e pertanto della sua induzione epatica di IGF1. [Watt HL et al]
Fino al momento in cui le cellule che costituiscono il primo aggregato tumorale di pochi millimetri non riescono a crearsi un proprio sistema di vasi sanguigni (Angiogenesi neoplastica), esse crescono con estrema lentezza e sono destinate a non superare le dimensioni di qualche millimetro, rimanendo allo stadio di "cancro in situ". L'espansione tumorale avviene solo quando il tumore realizza l'angiogenesi, riesce cioè a costruirsi una rete di vasi sanguigni per assicurarsi l'apporto di sostanze nutritive e l'eliminazione di scorie metaboliche. La letteratura ha documentato che tutti i passaggi dell'angiogenesi e le molecole che ad essa sinergicamente concorrono, (sia promotori dell'angiogenesi che fattori di crescita angiogenici) sono negativamente regolati dalla somatostatina e dai suoi analoghi e, anche se in misura minore, da tutti gli altri componenti del MDB. Se l'espansione neoplastica ha nell'angiogenesi un passaggio obbligato, e se l'angiogenesi è totalmente inibita dalla somatostatina, è ulteriormente chiarita e documentata la sua indicazione in tutti i tumori, in presenza o meno, di SSTR.
I Fenomeni di angiogenesi e neoangiogenesi, condizioni necessarie della progressione neoplastica, così come la cascata dei monociti, il rilascio paracrino di interleukina 8 e il contributo dei fattori di crescita (il cui sinergismo è essenziale per l'angiogenesi stessa), come il VEGF, TGF, IGF1, FGF, HGF, PDGF, costituiscono gli specifici bersagli molecolari negativamente regolati da Somatostatina e analoghi. L'inibizione dell'angiogenesi indotta dalla SST è sinergicamente potenziata da MLT, Retinoidi, vitamina D3, Vitamina E, Vitamina C, inibitori prolattinici e da componenti della matrice extracellulare. Anche le situazioni locali di ipossia/anossia e acidosi favoriscono l'angiogenesi, e in buona parte sono corrette dal miglioramento degli scambi emotissutali indotto dai componenti differenzianti del MDB.
Contemporaneamente, gli stessi effetti citostatico, antiproliferativo, antimetastatico della Somatostatina vengono sinergicamente incrementati dagli altri componenti del MDB.
Un ulteriore contributo viene fornito dalla somministrazione giornaliera di basse dosi [50-100 mg/die per os] di Ciclofosfamide (Endoxan). Questa posologia, oltre che ridurre drasticamente i noti effetti antiblastici/mielosoppressori, induce un marcato viraggio dei suoi meccanismi d'azione:
innesco della cascata apoptotica mitocondrio dipendente, azione antiangiogenetica mediante l'abbattimento del VEGF.
Ampiamente documentata è anche l'attività inibitoria della SST su un altro potente fattore di crescita mitogeno, EGF, attraverso molteplici meccanismi:
- Blocco del signaling dose-dipendente (Inibizione della fosforilazione tirosinica) del EGFR;
- Riduzione dell'espressione di EGFR e del suo ligando (EGF) nelle cellule tumorali;
- Abbattimento della concentrazione plasmatica di EGF.
La suddetta interferenza viene ulteriormente potenziata dalla concomitante somministrazione di MLT e Vitamina D3, la cui attività modulatoria nei confronti del fattore di crescita epidermico è ben nota. È stato osservato come i tumori al seno esprimano le sottoclassi recettoriali SSTR1, SSTR2, SSTR3, meno frequentemente SSTR5 [1,52]. Sebbene almeno nel 50% dei casi siano scintigraficamente riscontrabili, ulteriori indagini immunoistochimiche hanno rilevato la presenza di STTR anche in soggetti con esito scintigrafico negativo [Körner M et al.; Mezi S]. Pertanto la presenza di STTR e di recettori neuroendocrini in una rilevante percentuale di questi carcinomi, costituisce un'ulteriore indicazione razionale all'impiego della SST, peraltro già ampiamente giustificata dalla già citata regolazione negativa sul GH, dei GF GH-correlati, e dell'angiogenesi.
E' oramai assodato come gli ormoni sessuali giochino un ruolo chiave nella eziologia e progressione del tumore al seno, così come su svariate neoplasie ormono-dipendenti, quali il tumore prostatatico ed ovarico. Il principale meccanismo alla base di queste neoplasie è il risultato della prolungata stimolazione e sollecitazione ormonale, con gravi ripercussioni a lungo termine sulla crescita normale e funzione del tessuto bersaglio. Numerose evidenze sperimentali hanno dimostrato come sia il contributo degli ormoni sessuali, che gli GF da essi regolati, esercitino una profonda modulazione genica. La principale conseguenza di questa interferenza ormonale è rappresentata da una proliferazione cellulare incontrollata. L'impiego di basse dosi degli inibitori delle aromatasi di seconda generazione (Anastrozolo), già impiegata nella prassi clinica, combinati con MLT e SST; regola negativamente i processi di proliferazione ormono-dipendenti delle neoplasie Mammarie.
Numerosi investigazioni precliniche e sull'uomo hanno evidenziato i meccanismi d'azione della MLT. Essendo la molecola associata al Signaling Pathways dello sviluppo sia fisiologico epiteliale che neoplastico, tale indolo possiede sia la proprietà neutralizzare selettivamente i segnali proliferativi degli estrogeni che di modularne negativamente la loro biosintesi locale. L'impiego della MLT si estende su tutti istotipi di carcinoma mammario, data l'elevata distribuzione di membrana delle sottoclassi recettoriali MT1-MT2. Numerosi studi epidemiologici hanno altresì dimostrato che tra le molteplici cause predisponenti al carcinoma mammario vi è lo stravolgimento dei livelli fisiologici circadiani.
I meccanismi molecolari dell'indolo sono molteplici e riassumibili nei seguenti punti:
abbattimento dell'espressione genica dei recettori per gli estrogeni; nonché del signaling indotto dagli stessi,
interferenza sul metabolismo degli estrogeni,
modulazione epigenetica,
blocco dei processi di motilità cellulare, invasione e metastasi attraverso il blocco dell'espressione delle Metalloproteinasi
Nel dettaglio:
E' oramai assodato come gli ormoni sessuali giochino un ruolo chiave nella eziologia e progressione del tumore al seno, così come su svariate neoplasie ormono-dipendenti, quali il tumore prostatatico ed ovarico. Il principale meccanismo alla base di queste neoplasie è il risultato della prolungata stimolazione e sollecitazione ormonale, con gravi ripercussioni a lungo termine sulla crescita normale e funzione del tessuto bersaglio. Numerose evidenze sperimentali hanno dimostrato come sia il contributo degli ormoni sessuali, che gli GF da essi regolati, esercitino una profonda modulazione genica. La principale conseguenza di questa interferenza ormonale è rappresentata da una proliferazione cellulare incontrollata. L'impiego di basse dosi degli inibitori delle aromatasi di seconda generazione (Anastrozolo), già impiegata nella prassi clinica, combinati con MLT e SST; regola negativamente i processi di proliferazione ormono-dipendenti delle neoplasie Mammarie.
Numerosi investigazioni precliniche e sull'uomo hanno evidenziato i meccanismi d'azione della MLT. Essendo la molecola associata al Signaling Pathways dello sviluppo sia fisiologico epiteliale che neoplastico, tale indolo possiede sia la proprietà neutralizzare selettivamente i segnali proliferativi degli estrogeni che di modularne negativamente la loro biosintesi locale. L'impiego della MLT si estende su tutti istotipi di carcinoma mammario, data l'elevata distribuzione di membrana delle sottoclassi recettoriali MT1-MT2. Numerosi studi epidemiologici hanno altresì dimostrato che tra le molteplici cause predisponenti al carcinoma mammario vi è lo stravolgimento dei livelli fisiologici circadiani.
I meccanismi molecolari dell'indolo sono molteplici e riassumibili nei seguenti punti:
abbattimento dell'espressione genica dei recettori per gli estrogeni; nonché del signaling indotto dagli stessi,
interferenza sul metabolismo degli estrogeni,
modulazione epigenetica,
blocco dei processi di motilità cellulare, invasione e metastasi attraverso il blocco dell'espressione delle Metalloproteinasi
Nel dettaglio:
- La sottoregolazione dell'asse riproduttivo ipotalamico-ipofisario, con conseguente riduzione dei livelli circolanti di estrogeni gonadici, rappresenta uno dei principali meccanismi neuroendocrini diretto della MLT;
- La MLT è un modulatore selettivo del recettore per gli estrogeni può: agisce direttamente sulle cellule tumorali mammarie interferendo con l'attivazione recettoriale (ESR).
La letteratura ha pertanto confermato i meccanismi d'azione antineoplastici differenzianti e antiproliferativi, antiangiogenetici e antimetastatici di tutti i componenti del MDB, sia in vitro che in vivo.
Nel presente studio retrospettivo viene confermata la risposta obiettiva di neoplasie mammarie al trattamento concomitante delle suddette molecole biologiche.
Dato altrettanto degno di nota, la risposta obiettiva ottenuta in 15 casi senza fare riferimento ai consueti trattamenti poli-chemio-radio-terapici e/oi chirurgici; considerando che sia i fenomeni di recidiva che di metastasi post-operatorie attualmente sono la causa principale della mortalità correlata al cancro al seno, mentre l'impiego di dosi massicce di agenti antiblastici e di terapia radiante oltre che incrementare le mutazioni costituscono un terreno fertile per l'attivazione di fattori promuoventi la crescita e progressione dei tumori ormono-dipendenti.
L'impiego combinato delle molecole MDB ha favorito una sopravvivenza al primo anno di follow up totale, aumentando significativamente, la mediana di sopravvivenza per le pazienti in stadio avanzato di malattia.
Dei 5 casi sotto osservazione, attualmente si presenta una OS di oltre mesi, con una TPS di 13 mesi.
Il risultato obiettivo, in assenza di tossicità, mediante la riduzione progressiva, fino alla scomparsa, delle lesioni neoplastiche iniziali, nonché delle adenopatie ascellari, delle lesioni cerebrali, insieme al blocco di ogni disseminazione metastatica, evidenzia inequivocabilmente l'efficacia di questa
terapia ed è conforme ai positivi risultati preliminari già pubblicati sull'uso del MDB nei nelle patologie linfoproliferative e sui carcinomi polmonari in stadio avanzato e cervico facciali.
Il MDB, senza necessità di ricovero ospedaliero e/o di strutture di Day Hospital, in assenza di tossicità e senza ridurre minimamente l'attività lavorativa, ha evitato il trauma chirurgico, e i rilevanti effetti collaterali dei consueti protocolli chemioterapici e di terapia radiante.
Ne consegue pertanto come l'applicazione precoce e come terapia di prima linea del MDB, in un organismo non debilitato dagli effetti tossici, mutageni e immunodepressivi della chemio-radioterapia, abbia grandemente facilitato il risultati di risposta obiettiva, sopravvivenza e qualità di vita.
CONCLUSIONI
Il razionale teorico dell'impiego di molecole biologiche sulle neoplasie mammarie, supportato dai risultati clinici sopra riportati; comprova la logicità ed efficacia della concezione multiterapica del MDB, terapia biologica dei tumori: l'interazione sinergica dei suoi componenti asseconda ed esalta le reazioni vitali e l'omeostasi antitumorale per metterle in condizione di contrapporsi all'anarchia dei processi neoplastici del microambiente tumorale.
Sinteticamente, la terapia biologica si propone di contrastare la progressione del fenotipo
neoplastico attraverso:
Nel presente studio retrospettivo viene confermata la risposta obiettiva di neoplasie mammarie al trattamento concomitante delle suddette molecole biologiche.
Dato altrettanto degno di nota, la risposta obiettiva ottenuta in 15 casi senza fare riferimento ai consueti trattamenti poli-chemio-radio-terapici e/oi chirurgici; considerando che sia i fenomeni di recidiva che di metastasi post-operatorie attualmente sono la causa principale della mortalità correlata al cancro al seno, mentre l'impiego di dosi massicce di agenti antiblastici e di terapia radiante oltre che incrementare le mutazioni costituscono un terreno fertile per l'attivazione di fattori promuoventi la crescita e progressione dei tumori ormono-dipendenti.
L'impiego combinato delle molecole MDB ha favorito una sopravvivenza al primo anno di follow up totale, aumentando significativamente, la mediana di sopravvivenza per le pazienti in stadio avanzato di malattia.
Dei 5 casi sotto osservazione, attualmente si presenta una OS di oltre mesi, con una TPS di 13 mesi.
Il risultato obiettivo, in assenza di tossicità, mediante la riduzione progressiva, fino alla scomparsa, delle lesioni neoplastiche iniziali, nonché delle adenopatie ascellari, delle lesioni cerebrali, insieme al blocco di ogni disseminazione metastatica, evidenzia inequivocabilmente l'efficacia di questa
terapia ed è conforme ai positivi risultati preliminari già pubblicati sull'uso del MDB nei nelle patologie linfoproliferative e sui carcinomi polmonari in stadio avanzato e cervico facciali.
Il MDB, senza necessità di ricovero ospedaliero e/o di strutture di Day Hospital, in assenza di tossicità e senza ridurre minimamente l'attività lavorativa, ha evitato il trauma chirurgico, e i rilevanti effetti collaterali dei consueti protocolli chemioterapici e di terapia radiante.
Ne consegue pertanto come l'applicazione precoce e come terapia di prima linea del MDB, in un organismo non debilitato dagli effetti tossici, mutageni e immunodepressivi della chemio-radioterapia, abbia grandemente facilitato il risultati di risposta obiettiva, sopravvivenza e qualità di vita.
CONCLUSIONI
Il razionale teorico dell'impiego di molecole biologiche sulle neoplasie mammarie, supportato dai risultati clinici sopra riportati; comprova la logicità ed efficacia della concezione multiterapica del MDB, terapia biologica dei tumori: l'interazione sinergica dei suoi componenti asseconda ed esalta le reazioni vitali e l'omeostasi antitumorale per metterle in condizione di contrapporsi all'anarchia dei processi neoplastici del microambiente tumorale.
Sinteticamente, la terapia biologica si propone di contrastare la progressione del fenotipo
neoplastico attraverso:
a) L'inibizione della proliferazione neoplastica mediante i processi cellulari di apoptosi/necrosi e privazione dei ormoni e fattori di crescita cellulari;
b) Il contrasto della spiccata tendenza mutagena mediante l'attivazione diretta dei sistemi di riparazione del DNA, ed attraverso la riprogrammazione cellulare epigenetica.
c) Il blocco dell progressione neoplastica, mediante l'abbattimento della formazione dei binari ematici (Neoangiogenesi-Linfoangiogenesi) e dei fenomeni di motilità cellulare (migrazione), essenziali per la disseminazione neoplastica a lungo raggio;
d) La difesa dall'aggressione neoplastica attraverso il potenziamento delle difese naturali (immunità innata ed acquisita);
Il tumore infatti è da considerarsi come una deviazione afinalistica dell'omeostasi cellulare, per cui occorre riportare le reazioni biochimiche deviate verso la norma attraverso il potenziamento e la modulazione di tutti quei mezzi che la Fisiologia considera essenziali per la vita.
Il documentato sinergismo antiangiogenico di ogni componente del MDB, unitamente a quello antiproliferativo di somatostatina e inibitori prolattinici ed estrogenici e quello differenziante immunomodulante, trofico e omeostatico degli altri componenti del MDB, hanno conseguito questo risultato evitando da una parte la grave tossicità e danni a volte permanenti delle consuete terapie mediche del cancro, dall' altra incrementando notevolmente il PS e la qualità e durata della vita.
In conclusione, gli autori ritengono utile e doveroso segnalare tale indagine osservazionale per invitare la comunità scientifica ad un maggiore interesse. Riteniamo infatti che i pochi studi clinici fettuati sull' impiego multiterapico sinergico di queste molecole biologiche andrebbero approfonditi ed estesi alle varie patologie neoplastiche esaurientemente documentati e diffusi.
Futuri studi clinici randomizzati e a doppio cieco del suddetto trattamento sono ben auspicabili
REFERENCES
- Alvarez-García V, González A, Martínez-Campa C, Alonso-González C and Cos S (2013). Melatonin modulates aromatase activity and expression in endothelial cells. Oncol Rep. 29(5). 2058–64.
- Bernichtein S, Touraine P and Goffin V (2010). New concepts in prolactin biology. J Endocrinol. 206: 1–11.
- Cameron Smith M, Orlando C, Serio M and Maggi M (2003). Somatostatin receptors and breast cancer. J Endocrinol Invest. 26: 125–30.
- Carver KC, Piazza TM and Schuler LA (2010). Prolactin enhances insulin-like growth factor I receptor phosphorylation by decreasing its association with the tyrosine phosphatase SHP-2 in MCF-7 breast cancer cells. J Biol Chem. 285: 8003–12.
- Cescon DW, Ganz PA, Beddows S, Ennis M, Mills BK, and Goodwin PJ (2012). Feasibility of a randomized controlled trial of vitamin D vs. placebo in women with recently diagnosed breast cancer. Breast Cancer Res Treat. 134: 759–67.
- Chatzistamou I, Schally AV, Kiaris H. et al. (2004). Immunohistochemical detection of GHRH and its receptor splice variant 1 in primary human breast cancers. Eur J Endocrinol. 151: 391–6.
- Ciolino HP, Dai Z and Nair V (2011). Retinol inhibits aromatase activity and expression in vitro. J Nutr Biochem. 22: 522–6.
- Common Terminology Criteria for Adverse Events (CTCAE) and Common Toxicity Criteria (CTC) (http://ctep.can cer.gov/protocolDevelopment/electronic_applications/ctc.htm).
- Demicheli R, Abbattista A, Miceli R, Valagussa P, Bonadonna G (1996). Time distribution of the recurrence risk for breast cancer patients undergoing mastectomy: further support about the the concept of tumor dormancy. Breast Cancer Res Treat. 41(2). 177–85.
- Di Bella G: The Di Bella Method (DBM)(2010). Neuro Endocrinol Lett. 31: 1–42.
- Di Bella G and Colori B (2012). The Di Bella Method (DBM) improved survival, objective response and performance status in a retrospective observational clinical study on 23 tumours of the head and neck. Neuro Endocrinol Lett. 33: 249–56.
- Faupel-Badger JM, Sherman ME, Garcia-Closas M et al. (2010). Prolactin serum levels and breast cancer: relationships with risk factors and tumour characteristics among pre- and postmenopausal women in a population-based case-control study from Poland. Br J Cancer. 103: 1097–102.
- Fisher B, Gunduz N, Coile J, Rudock C, Saffer E. (1989). Presence of a growth-stimulating factor in serum following primary tumour removal in mice. Cancer Res. 49: 1996–2001.
- Frank SJ (2008). Mechanistic aspects of crosstalk between GH and PRL and ErbB receptor family signaling. J Mammary Gland Biol Neoplasia. 13: 119–29.
- Frati A, Antoine M, Rodenas A, Gligorov J, Rouzier R and Chéreau E (2011). Somatostatin in breast cancer. Ann Biol Clin (Paris). 69: 385–91.
- Fulan H, Changxing J, Baina WY et al. (2011). Retinol, vitamins A, C, and E and breast cancer risk: a meta-analysis and metaregression. Cancer Causes Control. 22: 1383–96.
- Fürstenberger G, Morant R and Senn HJ (2003). Insulin-like growth factors and breast cancer. Onkologie. 26: 290–4.
- Gebre-Medhin M, Kindblom LG, Wennbo H, Törnell J and Meis-Kindblom JM (2001). Growth hormone receptor is expressed in human breast cancer. Am J Pathol. 158: 1217–22.
- Girgert R, Hanf V, Emons G and Gründker C (2009). Membranebound melatonin receptor MT1 down-regulates estrogen responsive genes in breast cancer cells. J Pineal Res. 47: 23–31.
- Haines E, Minoo P, Feng Z et al. (2009). Tyrosine phosphorylation of Grb2: role in prolactin/epidermal growth factor cross talk in mammary epithelial cell growth and differentiation. Mol Cell Biol. 29: 2505–20.
- Harvey PW (2012). Hypothesis: prolactin is tumorigenic to human breast: dispelling the myth that prolactin-induced mammary tumors are rodent-specific. J Appl Toxicol. 32: 1–9.
- He Y, Yuan XM, Lei P et al. (2009). The antiproliferative effects of somatostatin receptor subtype 2 in breast cancer cells. Acta Pharmacol Sin. 30: 1053–9
- Hill SM, Blask DE, Xiang S et al. (2011). Melatonin and associated signaling pathways that control normal breast epithelium and breast cancer. J Mammary Gland Biol Neoplasia. 16: 235–45.
- Hofland LJ, van der Burg B, van Eijck CH, Sprij DM et al. (1995). Role of tumor-derived fibroblasts in the growth of primary cultures of human breast-cancer cells: effects of epidermal growth factor and the somatostatin analogue octreotide. Int J Cancer. 60: 93–9.
- Jacobson EM, Hugo ER, Tuttle TR, Papoian R and Ben-Jonathan N (2010). Unexploited therapies in breast and prostate cancer: blockade of the prolactin receptor. Trends Endocrinol Metab. 21: 691–8.
- Kamenicky P, Lombès M and Chanson P (2010). [New insights in growth hormone physiology and pathophysiology]. Ann Endocrinol (Paris). 71: S25–32.
- Kaulsay KK, Zhu T, Bennett W, Lee KO and Lobie PE (2001). The effects of autocrine human growth hormone (hGH) on human mammary carcinoma cell behavior are mediated via the hGH receptor. Endocrinology. 142: 767–77.
- Kim KJ, Choi JS, Kang I et al. (2013). Melatonin suppresses tumor progression by reducing angiogenesis stimulated by HIF-1 in a mouse tumor model. J Pineal Res. 54(3): 264–70.
- Knower KC, To SQ, Takagi K (2012). Melatonin suppresses aromatase expression and activity inbreast cancer associated fibroblasts. Breast Cancer Res Treat. 132: 765–71.
- Kumar U, Grigorakis SI, Watt HL et al. (2005). Somatostatin receptors in primary human breast cancer: quantitative analysis of mRNA for subtypes 1--5 and correlation with receptor protein expression and tumor pathology. Breast Cancer Res Treat. 92: 175–86.
- Laban C, Bustin SA and Jenkins PJ (2003). The GH-IGF-I axis and breast cancer. Trends Endocrinol Metab 14: 28–34.
- Lagadec C, Vlashi E, Della Donna L, et al. (2012). Radiationinduced reprogramming of breast cancer cells. Stem Cells. 30: 833.
- Lee LT, Schally AV, Liebow C et al. (2008). Dephosphorylation of cancer protein by tyrosine phosphatases in response to analogs of luteinizing hormone-releasing hormone and somatostatin. Anticancer Res. 28: 599–605.
- Li X, Huang Y, Jiang J and Frank SJ (2011). Synergy in ERK activation by cytokine receptors and tyrosine kinase growth factor receptors. Cell Signal. 23: 417–24.
- Lissoni P, Rovelli F, Malugani F, Bucovec R, Conti A, Maestroni GJ (2001). Anti-angiogenic activity of melatonin in advanced cancer patients. Neuro Endocrinol Lett. 22: 45–7.
- Loven D, Hasnis E, Bertolini F and Shaked Y (2013). Low-dose metronomic chemotherapy: from past experience to new paradigms in the treatment of cancer. Drug Discov Today. 18: 193–201.
- Margheri M. et al. (2012). Combined effects of melatonin and all-trans retinoic acid and somatostatin on breast cancer cell proliferation and death: molecular basis for the anticancer effect of these molecules. Eur J Pharmacol. 68: 34–43.
- Marie P, Clunet, J. (1910). Fréquences des métastases viscérales chez les souris cancéreuses aprčs ablation chirurgicale de leur tumeur. Bull. Assoc. Fr. Etud. Cancer. 3: 19–23.
- Mehta RG, Peng X, Alimirah F et al. (2012). Vitamin D and breast cancer: Emerging concepts. Cancer Lett. pii: S0304-3835(12)00639-8. doi: 10.1016/j.canlet.2012.10.034. [Epub ahead of print].
- Milewicz T, Ryś J, Wójtowicz A, et al. (2011). Overexpression of P53 protein and local hGH, IGF-I, IGFBP-3, IGFBP-2 and PRL secretion by human breast cancer explants. Neuro Endocrinol Lett. 32: 328–33.
- Moody TW, Chan D, Fahrenkrug J and Jensen RT (2003). Neuropeptides as autocrine growth factors in cancer cells. Curr Pharm Des. 9: 495–509.
- Naumov GN, MacDonald IC, Weinmeister PM, Kerkvliet N et al. (2002). Persistence of solitary mammary carcinoma cells in a secondary site: a possible contributor to dormancy. Cancer Res. 62: 2162–2168.
- Norsa A and Martino V (2007). Somatostatin, retinoids, melatonin, vitamin D, bromocriptine, and cyclophosphamide in chemotherapy-pretreated patients with advanced lung adenocarcinoma and low performance status. Cancer Biother Radiopharm. 22: 50–5.
- Norsa A and Martino V (2006). Somatostatin, retinoids, melatonin, vitamin D, bromocriptine, and cyclophosphamide in advanced non-small-cell lung cancer patients with low performance status. Cancer Biother Radiopharm. 21: 68–73.
- Oprea-Ilies G, Haus E, Sackett-Lundeen L et al. (2012). Expression of melatonin receptors in triple negative breast cancer (TNBC) in African American and Caucasian women: relation to survival. Breast Cancer Res Treat. 137(3). 677–87.
- Ostendorf GM (2012). High dosage vitamin C in breast cancer? Versicherungsmedizin. 64: 85.
- Patrick T. et al. (2000). New Guidelines to Evaluate the Response to Treatment in Solid Tumors. Journal of the National Cancer Institute, Vol. 92.
- Picotto G, Liaudat AC, Bohl L and Tolosa de Talamoni N (2012). Molecular aspects of vitamin D anticancer activity. Cancer Invest. 30: 604–14.
- Perry JK, Mohankumar KM, Emerald BS, Mertani HC and Lobie PE (2008). The contribution of growth hormone to mammary neoplasia. J Mammary Gland Biol Neoplasia. 13: 131–45.
- Pollak M (1997). The potential role of somatostatin analogues in breast cancer treatment. Yale J Biol Med. 70: 535–9.
- Proietti S, Cucina A, Reiter RJ and Bizzarri M (2010). Molecular mechanisms of melatonin’s inhibitory actions on breast cancers. Cell Mol Life Sci. 70(12). 2139–57.
- Proietti S, Cucina A, D’Anselmi F, et al. (2011). Melatonin and vitamin D3 synergistically down-regulate Akt and MDM2 leading to TGFβ-1-dependent growth inhibition of breast cancer cells. J Pineal Res. 50: 150–8.
- Raccurt M, Lobie PE, Moudilou E, et al. (2002). High stromal and epithelial human gh gene expression is associated with proliferative disorders of the mammary gland. J Endocrinol. 175: 307–18.
- Rasmussen LM, Frederiksen KS, Din N, Galsgaard E, Christensen L, Berchtold MW and Panina S (2010). Prolactin and oestrogen synergistically regulate gene expression and proliferation of breast cancer cells. Endocr Relat Cancer. 17: 809–22.
- Rögelsperger O, Wlcek K, Ekmekcioglu C et al. (2011). Melatonin receptors, melatonin metabolizing enzymes and cyclin D1 in human breast cancer. J Recept Signal Transduct Res. 31: 180–7.
- Ruscica M, Arvigo M, Steffani L, Ferone D and Magni P (2012). Somatostatin, somatostatin analogs and somatostatin receptor dynamics in the biology of cancer progression. Curr Mol Med. 13(4): 555–71.
- Sackett DL et al. (1996). “Evidence based medicine: what it is and what it isn’t”. BMJ. 312(7023): 71–2.
- Salvatori L, Ravenna L, Caporuscio F, et al. (2011). Action of retinoic acid receptor on EGFR gene transactivation and breast cancer cell proliferation: Interplay with the estrogen receptor. Biomed Pharmacother. 65: 307–12.
- Sanchez-Barcelo EJ, Mediavilla MD, Alonso-Gonzalez C and Reiter RJ (2012). Melatonin uses in oncology: breast cancer prevention and reduction of the side effects of chemotherapy and radiation. Expert Opin Investig Drugs. 21: 819–31.
- Sanchez-Barcelo EJ, Mediavilla MD, Alonso-Gonzalez C and Rueda N (2012). Breast cancer therapy based on melatonin. Recent Pat Endocr Metab Immune Drug Discov. 6: 108–16.
- Seitz S, Buchholz S, Schally AV et al. (2013). Targeting triplenegative breast cancer through the somatostatin receptor with the new cytotoxic somatostatin analogue AN-162 (AEZS-124). Anticancer Drugs. 24: 150–157.
- Schally AV, Comaru-Schally AM, Nagy A et al. (2001). Hypothalamic hormones and cancer. Front Neuroendocrinol 22: 248–91.
- Schally AV and Nagy A (2003). New approaches to treatment of various cancers based on cytotoxic analogs of LHRH, somatostatin and bombesin. Life Sci. 72: 2305–20.
- Schally AV (2008). New approaches to the therapy of various tumors based on peptide analogues. Horm Metab Res. 40: 315–22.
- Siriwardana G, Bradford A, Coy D and Zeitler P (2006). Autocrine/paracrine regulation of breast cancer cell proliferation by growth hormone releasing hormone via Ras, Raf, and mitogenactivated protein kinase. Mol Endocrinol. 20: 2010–9.
- Smith J, Axelrod D, Singh B and Kleinberg D (2011). Prevention of breast cancer: the case for studying inhibition of IGF-1 actions. Ann Oncol. 22: i50–2.
- Sogno I, Venè R, Sapienza C, Ferrari N, Tosetti F, Albini A (2009). Anti-angiogenic properties of chemopreventive drugs: fenretinide as a prototype. Recent Results Cancer Res. 181: 71–6.
- Suhail N, Bilal N, Khan HY et al. (2012). Effect of vitamins C and E on antioxidant status of breast- cancer patients undergoing chemotherapy. J Clin Pharm Ther. 37: 22–6.
- Sun Y, Campisi J, Higano C et al. (2012). Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 8: 1359–1368.
- Tang XH and Gudas LJ (2011). Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol. 6: 345–64.
- Tyzzer EE (1913). Factors in the production and growth of tumor metastases. Cancer Res. Clin. Oncol. 13: 546–547.
- Tejeda M, Gaál D, Hullán L, Hegymegi-Barakonyi B and Kéri G (2006). Evaluation of the antitumor efficacy of the Somatostatin structural derivative TT-232 on different tumor models. Anticancer Res. 26: 3477–83.
- Todisco M, Casaccia P and Rossi N (2001). Cyclophosphamide plus somatostatin, bromocriptin, retinoids, melatonin and ACTH in the treatment of low-grade non-Hodgkin’s lymphomas at advanced stage: results of a phase II trial: Cancer Biother Radiopharm 16: 171–7.
- Voss MJ, Möller MF, Powe DG et al. (2011). Luminal and basallike breast cancer cells show increased migration induced by hypoxia, mediated by an autocrine mechanism. BMC Cancer. 2: 158.
- Wang J, Xiao X, Zhang Y et al. (2012). Simultaneous modulation of COX-2, p300, Akt, and Apaf-1 signaling by melatonin to inhibit proliferation and induce apoptosis in breast cancer cells. J Pineal Res. 53: 77–90.
- Watt HL, Kharmate GD and Kumar U (2009). Somatostatin receptors 1 and 5 heterodimerize with epidermal growth factor receptor: agonist-dependent modulation of the downstream MAPK signalling pathway in breast cancer cells. Cell Signal. 21: 428–39.
- Watt HL, Kharmate G and Kumar U (2008). Biology of somatostatin in breast cancer. Mol Cell Endocrinol. 286: 251–61.
- Wu ZS. et al. (2011). Tumor expression of human growth hormone and human prolactin predict a worse survival outcome in patients with mammary or endometrial carcinoma. J Clin Endocrinol Metab. 96: E1619–29.
- Xu J, Sun D, Jiang J, Deng L, Zhang Y et al. (2012). The Role of Prolactin Receptor in GH Signaling in Breast Cancer Cells. Mol Endocrinol. 27(2): 266–79.
- Zhang Y, Zhang H, Wang X et al. (2012). The eradication of breast cancer and cancer stem cells using octreotide modified paclitaxel active targeting micelles and salinomycin passive targeting micelles. Biomaterials. 33: 679–91.
- Zheng RH, Guan XX, Zhang XP, et al. (2010). [The correlation between the expression of PRL-R and ER/PR in breast cancer]. Nan Fang Yi Ke Da Xue Xue Bao. 30: 596–8.