Il Metodo Di Bella. Casistica su 553 casi
Pubblicato il 18/09/2014
Il Metodo Di Bella ( MDB)
Giuseppe Di Bella Di Bella Foundation, Bologna, Italy.
Correspondence to: Giuseppe Di Bella, MD. PhD. Via Marconi n 51, Bologna 40122 ▪ Italy
tel: +39-051-239662; +39-051-230369; fax: +39-051-2961283
e-mail: posta@giuseppedibella.it
Key words: metodo di bella, somatostatina, octreotide, melatonina, retinoidi, vitamine E, C, D3, Bromocriptina, Cabergolina, Condroitinsolfato, calcio
Abstract
Obiettivi: scopo del MDB è superare l’elevata tossicità e bassa efficacia delle attuali terapie mediche del cancro.
Metodo: con MLT, Retinoidi ,vitamine E, D3, C, componenti della ECM, il MDB potenzia quei mezzi che la Fisiologia considera essenziali per la vita. Queste molecole esercitano sinergicamente, un ruolo differenziante e antiproliferativo. Con Somatostatina e/o analoghi il MDB inibisce l’oncogenesi, regolando negativamente molecole mitogene come il GH e i fattori di crescita GH dipendenti. Con Cabergolina e/o Bromocriptina regola negativamente la Prolattina, ormone mitogeno ubiqitario. Il MDB prevede minimali dosaggi apoptotici, non citotossici e non mutageni di Ciclofosfamide o Oncocarbide, la cui tollerabilità è esaltata dalla MLT e dalle vitamine del MDB.
Risultati: sono riportati i risultati preliminari dello studio osservazionale retrospettivo su 553 pazienti trattati con MDB, esaminati e certificati da tre medici nominati Consulenti Tecnici di Ufficio dalla Procura di Lecce , in Italia. Questi dati documentano che il MDB ha conseguito un evidente miglioramento della qualità di vita ed un sensibile incremento delle mediane di sopravvivenza per ogni patologia e stadio rispetto ai dati reperibili in letteratura relativi alla chemioterapia e/o anticorpi monoclonali.Il risultato è stato conseguito in totale assenza dei noti e rilevanti effetti tossici della chemioterapia e anche se in misura minore rispetto alla chemio, degli anticorpi monoclonali .
Sono anche documentate le cause invalidanti che hanno totalmente destituito di ogni credibilità scientifica la sperimentazione del MDB effettuata in Italia nel 1998.
Conclusioni: ho ritenuto utile informare la comunità scientifica sul razionale, meccanismo d’azione, basi scientifiche e riscontri clinici del MDB, per invitare ad un maggior interesse sulle prospettive aperte dal MDB mediante innovative formulazioni delle vitamine e della MLT e l’impiego di molecole biologiche ad elevata efficacia antitumorale e bassa tossicità come la Somatostatina e analoghi.
Abbreviazioni
ATRA – All Trans Retinoic Acid
CCK – Cholecystokinin
C.M. – Chemiotassi dei Monociti GH indotta
CTU – Consulente tecnico d’ufficio
MDB – Metodo Di Bella
EGF – Epidermal Growth Factor
EGFR – Epidermal Growth Factor Receptor
FGF – Fibroblastic Growth Factor
G – Gastrin
GF – Growth Factor
2GH – Growth Hormone
GHR – Growth Hormone Receptor
HGF – Hepatocyte Growth Factor,
IGF1-2 – Insulin-like Growth Factor 1-2
IGFR – Insulin-like Growth Factor Receptor
IL8 – Interleuchin 8
MRI – Magnetic Resonance Imaging
MLT – Melatonin
NGF – Nerve Growth Factor
NHL – Non-Hodgkin’s Lymphoma
NOSe – Ossido-Nitrico-Sintasi endoteliale
PDGF – Platelet-Derived Growth Factor
PET – Positron Emission Tomography
PG2 – Prostaglandin 2
SSN – Servizio Sanitario Nazionale
SST – Somatostatina
SSTR – Somatostatin Receptor
TGF – Transforming Growth Factor
TRK – Tyrosine-kinase
VEGF – Vascular Endothelial Growth Factor
VIP – Vasoactive Intestinal Peptide
Sono anche documentate le cause invalidanti che hanno totalmente destituito di ogni credibilità scientifica la sperimentazione del MDB effettuata in Italia nel 1998.
Conclusioni: ho ritenuto utile informare la comunità scientifica sul razionale, meccanismo d’azione, basi scientifiche e riscontri clinici del MDB, per invitare ad un maggior interesse sulle prospettive aperte dal MDB mediante innovative formulazioni delle vitamine e della MLT e l’impiego di molecole biologiche ad elevata efficacia antitumorale e bassa tossicità come la Somatostatina e analoghi.
Abbreviazioni
ATRA – All Trans Retinoic Acid
CCK – Cholecystokinin
C.M. – Chemiotassi dei Monociti GH indotta
CTU – Consulente tecnico d’ufficio
MDB – Metodo Di Bella
EGF – Epidermal Growth Factor
EGFR – Epidermal Growth Factor Receptor
FGF – Fibroblastic Growth Factor
G – Gastrin
GF – Growth Factor
2GH – Growth Hormone
GHR – Growth Hormone Receptor
HGF – Hepatocyte Growth Factor,
IGF1-2 – Insulin-like Growth Factor 1-2
IGFR – Insulin-like Growth Factor Receptor
IL8 – Interleuchin 8
MRI – Magnetic Resonance Imaging
MLT – Melatonin
NGF – Nerve Growth Factor
NHL – Non-Hodgkin’s Lymphoma
NOSe – Ossido-Nitrico-Sintasi endoteliale
PDGF – Platelet-Derived Growth Factor
PET – Positron Emission Tomography
PG2 – Prostaglandin 2
SSN – Servizio Sanitario Nazionale
SST – Somatostatina
SSTR – Somatostatin Receptor
TGF – Transforming Growth Factor
TRK – Tyrosine-kinase
VEGF – Vascular Endothelial Growth Factor
VIP – Vasoactive Intestinal Peptide
Introduzione
Si descrivono il razionale del MDB, le finalità, i componenti, le basi biochimiche, fisiologiche e i meccanismi d’azione di biologia molecolare. Si documentano la tollerabilità, i riscontri clinici, la conferma in letteratura dell’efficacia antitumorale di ogni singolo componente del MDB, esaltata dall’effetto sinergico fattoriale. Si evidenziano le gravi e numerose anomalie che hanno totalmente delegittimato la sperimentazione del MDB del 1998. Si riportano i positivi risultati ottenuti dallo studio osservazionale retrospettivo su pazienti trattati con MDB.
Gli attuali dati della letteratura sulla chemioterapia documentano un’elevata tossicità e una percentuale di mortalità denunciata anche da un’agenzia della Reuters Healt [Wesport,CT 2001-05-17]: “Unexspected high mortality rated associated with chemoterapy regimen...”. Il dato è confermato da uno studio sui protocolli chemioterapici delle malattie linfoproliferative (Atra et al, 1998) che riporta l’undici per cento di decessi, non causati dal tumore ma unicamente da chemioterapia .
Oggi la sopravvivenza dei malati di tumore, è essenzialmente dovuta alla chirurgia, molto meno alla radioterapia, ed è del 29% a 5 anni (Richards et al, 2000). Di questo 29% solo il 2,1%-2,5% è dovuto alla chemio, (Morgan et al, 2005). Questa fondamentale pubblicazione si basa su 14 anni di osservazione, 225000 pazienti, 22 varietà tumorali, per accertare il reale contributo della chemio al raggiungimento dei 5 anni di sopravvivenza. La sola chemioterapia, senza chirurgia, consente pertanto solo al 2,1% ▪ 2,5% di raggiungere i 5 anni, dopo i quali è stato accertato che metà di questi pazienti sopravvissuti cinque anni, nel lungo termine muore per tumore (Lopez et al, 1998).
Dai recenti congressi dell’ American Society of Clinical Oncology, emerge chiaramente il dato che nei tumori solidi gli anticorpi monoclonali consentono in media un incremento della sopravvivenza di circa due mesi, e solo in rari casi ,con o senza abbinamento alla chemioterapia, si raggiungono o superano i quattro mesi.
In base a questi dati riportati dalla letteratura, abbiamo ritenuto opportuno proporre i nuovi percorsi terapeutici biologici, fisiologici, razionali,del MDB di maggiore efficacia e minore tossicità.
Metodo
La terapia (componenti del MDB)
Il MDB è così formulato:
A) Un modulo fisso, che agisce sui denominatori comuni a tutti i tumori.
B) Un modulo variabile, utilizzato in base alla peculiarità di specifiche neoplasie.
Il MDB è così formulato:
A) Un modulo fisso, che agisce sui denominatori comuni a tutti i tumori.
B) Un modulo variabile, utilizzato in base alla peculiarità di specifiche neoplasie.
MODULO FISSO
Vede l’utilizzo dei seguenti componenti
1)Acido Tutto-Trans Retinoico
Axeroftolo palmitato
Betacarotene
Alfatocoferile acetato
Queste molecole sono miscelate sotto forma di soluzione, formulazione che consente la massima biodisponibiltà, in questi rapporti:
Acido Tutto-Trans Retinoico 0, 5 gr
Axeroftolo palmitato 0,5 gr
Betacarotene 1 gr
Alfatocoferile acetato 1000 gr
Si calcola la dose giornaliera in base ai decimali del peso corporeo, per cui un adulto di 70 Kg può assumere 7 grammi di Soluzione 3 volte al dì
2) Melatonina compresse, nella formulazione del Prof. Di Bella, chimicamente complessata nel seguente rapporto: Melatonina 12% 5Adenosina 51%, Glicina 37% ,somministrata in compresse, in dosaggi dai 20 ai 60 mg al giorno
3) Bromocriptina compresse, 2,5 mg al giorno frazionata in ½ compressa mattino e sera
4) Cabergolina compresse da 0,5 mg. Può essere usata sia in sostituzione alla Bromocriptina, somministrando ½ compressa 2 volte la settimana
5) Diidrotachisterolo, Vit D3 di sintesi, 10 gocce prima del pasto con la soluzione dei retinoidi 3 volte al dì
6) Condroitinsolfato, bustine da 800 mg una mattino e sera diluite in acqua
7) Ciclofosfamide compresse 50 mg, da una a due al dì
8) Idrossiurea compresse 500 mg , da una a due al dì in alternativa alla ciclofosfamide
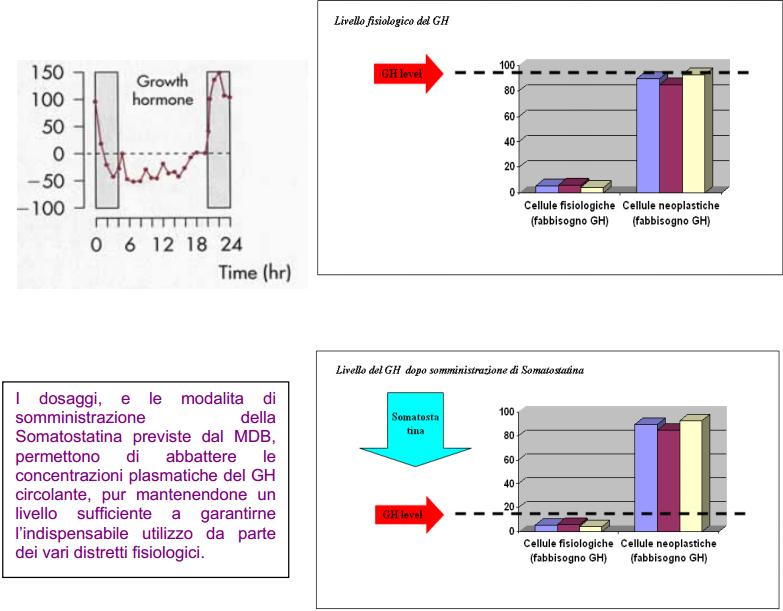
9) Somatostatina, peptide di 14 aminoacidi, alla dose di 3 mg giornalieri, iniettata la sera dopo cena, lentamente sottocute oppure in vena, con siringa temporizzata regolata a 12 ore (è indispensabile la somministrazione serale in quanto coincidente col picco notturno dell’increzione del GH e dei fattori di crescita GH-dipendenti)
10) Octreotide, peptide di 8 aminoacidi, in fiale da 1 mg/die con analoga modalità (diversamente si può usare la formulazione ritardo di Octreotide per via intramuscolo agli stessi dosaggi )
11) Vit. C, alle dosi di 2-4 grammi al dì, per bocca
12) Calcio, alle dosi di 2 grammi al giorno, per bocca
MODULO VARIABILE
Prevede il possibile utilizzo dei seguenti componenti:
- Inibitori degli androgeni nei tumori ormono-dipendenti dell’uomo
- Inibitori degli estrogeni nei tumori ormono-dipendenti della donna (ad esclusione del Tamoxifene per le possibili complicazioni tromboemboliche e di induzione neoplastica)
- ACTH di sintesi, tetracosactide esacetato, polipeptide di 24 aminoacidi, con le stesse indicazioni dei cortisonici, in sostituzione di questi per il miglior rapporto tra tollerabilità ed efficacia. Nel dosaggio di 1 mg settimanale intramuscolo, compatibilmente con la pressione, la glicemia, l’omeostasi elettrolitica.
- Glifosina [N,N-bis(fosfometil)glicina] in capsule da 200 mg, 2 volte al dì, per bocca, nelle neoplasie primitive o secondarie del tessuto osteocartilagineo
- Isoniazide, una capsula da 200 mg al giorno, nei carcinomi polmonari o della vescica.
- Anidrometilencitrato-esametilentetramina, soluzione acquosa al 10%, un cucchiaio 3 volte al dì (ai pasti) negli epatocarcinomi HCV correlati
- Dibromomannitolo nelle trombocitemie. Utilizzato in microdosi di circa 30 - 40 mg, a giorni alterni
- Albumina umana al 20-25% in 50 ml in vena nelle disprotidemie e nei versamenti pleurici e ascitici.
- Lenograstim, oppure Filgrastim, fattori di crescita dei globuli bianchi, nelle leucopenie, somministrati sottocute o in vena, fino al recupero dei valori fisiologici.
- Eritropoietina negli stati anemici, associata, nelle anemie sideropeniche, a ferro per os o in vena, e folati.
- Lisozima in compresse da 500 mg, alle dosi di 5-6 grammi al dì, nelle concomitanti patologie infettive, per l’azione antivirale e antibatterica, antiprotozoaria atossica, e per il potenziamento immunitario .
- mmunoglobuline, 5 ml intramuscolo al dì, (nelle patologie infettive concomitanti) per il potenziamento immunitario.
- Acido Fenil Chinolin Carbonico in dosi di 1000 mg per bocca, mattino e sera il 1° giorno, un grammo una sola volta al dì il 2° e 3° giorno, accompagnati in questi 3 giorni da 2 litri di acqua oligominerale. L’Acido Fenil Chinolin Carbonico, con questa posologia, normalizza rapidamente il tasso uricemico con profilo tossicità/vantaggi nettamente più favorevole degli inibitori delle monoamino-ossidasi comunemente impiegati in queste situazioni. Questi inibitori cronicamente somministrati, bloccando catene enzimatiche di alta dignità funzionale, possono indurre altre gravi patologie .
- Taurina in capsule da 200 mg, due volte al dì, per attivare l’effetto coleretico, colagogo e gli scambi emotissutali.
- Seleniometionina in capsule da 40 microgrammi, due volte al dì, con azione antiossidante e antiradicali liberi.
- Calciolevofolinato capsule 22 mg una al dì, sia in funzione differenziante che mieloprotettiva.
Meccanismi d’azione antitumorale dei componenti del MDB
Retinoidi, Betacarotene
- Esercita effetto protettivo sulle membrane cellulari (Di Bella, 1998)
- Diminuisce la perossidazione lipidica e aumenta il Glutatione (Basu et al., 2000)
- Esercita un effetto antiproliferativo diretto(indipendentemente dalla conversione in ATRA), sulle cellule tumorali, ne sopprime in modo significativo sia la mobilità (misurata mediante tetrazolium “MTT”), che la sintesi del DNA(controllata attraverso la captazione di -3H-timidina) e la proliferazione cellulare(misurata attraverso il conteggio delle cellule) (Onogi et al., 1998)
Vit. A ( Axeroftolo o retinolo )
- Provoca la morte della cellula neoplastica per apoptosi, attraverso l’attivazione di enzimi cellulari proteolitici, le Caspasi, e la degradazione del fattore della trascrizione generale Sp-1 [Piedrafita et al., 1997]
Ac retinoico ( All Trans Retinoic Acid –A.T.R.A.)
- Agisce ridifferenziando i blasti e le cellule tumorali [Hassan et al., 1990]
- Induce la sintesi di leucotriene C4 [Abe et al., 2003]
- Sopprime la trascrizione genica di fattori oncogeni e promuove l’effetto antiproliferativo [Arnold et al., 1994]
- Esercita azione anti-angiogenetica [Majewski et al., 1994]
- Diminusce la densità microvascolare del midollo osseo, nelle leucemie, e della densità del punto caldo. Interrompe la produzione di VEGF da parte delle cellule NB4, sopprimendo l’angiogenesi [Kini et al., 2001]
- Arresta lo sviluppo cellulare associato all’aumento dei livelli d’interferone 1 [IRF-1] con attivazione di p21WAF1 [Arany et al., 2003].
- Attiva l’apoptosi, col concorso di IRF-1 e STAT1, mediante la caspasi 1 [Arany et al., 2003].
- Arresta la progressione del ciclo cellulare [Wu et al., 2009].
- Induce l’arresto del ciclo cellulare in G0/G1 [Wu et al., 2009].
- Induce l’espressione di p21 WAF1/CIP 1, mediante percorsi sia dipendenti, che indipendenti da p 53 [Wu et al., 2009].
- Inibisce, nelle cellule tumorali, l’attività della proteina-1 attivatrice [AP-1] mediante il suo recettore RAR-alfa e attiva la soppressione dell’espressione di cJun e cFos [Wu et al., 2009].
- Sinergizza l’effetto di Bcl-2, sia sull’arresto della crescita, che sull’espressione del gene p21 [Chou et al., 2000].
- Impedisce l’invasione delle cellule del cancro del colon e diminuisce l’espressione del matrilysin [Adachi et al., 2001].
- Provoca, nelle cellule neoplastiche, cambiamenti morfologici e biochimici come il restringimento della membrana, la condensazione della cromatina e la spaccatura del DNA, caratteristiche tipiche delle cellule in corso di apoptosi [Lee et al., 2008] .
- Attiva tramite RAR-beta un netto incremento di proteine c-myc e Bax, che portano maggiore suscettibilità all’apoptosi [Lee et al., 2008].
- Diminuisce il potenziale di proliferazione neoplastica e ha un ruolo importante nella differenziazione, apoptosi e adesione cellulare [Voigt et al., 2000].
- Rende le cellule neoplastiche particolarmente sensibili ai chemioterapici, inducendo anche un aumento della comunicazione intercellulare negli spazi di giunzione [Carystinos et al., 2001].
- Riduce il livello della proteina silicea fibrillare gliale e la sintesi del DNA, e induce percorsi apoptotici, dimostrando un notevole sinergismo e potenziamento dell’efficacia col TNF-alfa mediante aumento dei recettori di p55 TNF [Chambaut-Guerin et al., 2000].
- Induce un gene, l’autotaxin [ATX], che decodifica un fattore di stimolazione della motilità del tumore [Duffner Beattie et al., 2001].
- Induce differenziazione neurotica con estesa crescita dei neuriti, diminuzione dell’oncoproteina n-Myc e del mRNA di Gap-43. Esercita l’effetto antiproliferativo attraverso l’incremento della chinasi A della proteina di tipo II/RII beta e chinasi A della proteina W [Kim et al., 2009].
- Differenzia le cellule neoplastiche attraverso il suo effetto sulle fosfolipasi A2, Ca2+-dipendenti [Antony et al., 2009].
- Riduce l’espressione di VnR, correlata all’organizzazione della fibronectina e all’adesione ed espansione cellulare [Baroni et al., 2003].
- Riduce l’inibizione chimicamente indotta di RAR Beta bloccando il ciclo cellulare in fase G1 [Song et al., 2001].
Vitamina E
- inibisce la crescita di varie linee cellulari tumorali, come:
- Cellule di carcinoma della prostata [Israel et al.,2000; Yu et al,.2002; Zhang et al., 2002]
- Cellule di carcinoma del seno [Yu et al., 1999; Pussinen et al. 2000; Yu et al.,1999]
- Cellule di carcinoma del polmone [Neuzil et al. 2001]
- Cellule di carcinoma della parotide [Prasad et al., 1996]
- Cellule di carcinoma dello stomaco [Rose et al., 2001; Wu et al. 2002]
- Cellule di carcinoma del colon [Neuzil et al., 2001]
- Cellule di carcinoma del pancreas [Heisler et al., 2000]
- Cellule di carcinoma squamoso orale [ Elattar et al.,1999]
- Cellule di melanoma [Prasad et al., 1990]
- Cellule di neuroblastoma [Prasad et al., 2003]
- Cellule di glioma [Prasad et al. 2003]
- Cellule leucemiche [Yamamoto et al., 2000]
- Cellule di linfoma [Turley et al., 1995; Yu et al., 1997; Dalen et al., 2003]
- A basse dosi induce differenziazione e inibizione della proliferazione tumorale; a concentrazioni maggiori induce apoptosi [Prasad et al., 2003]
- Soppressione della crescita tumorale [Prasad 2003]
- Attività apoptotica e/o citostatica di cellule del carcinoma del seno [Malafa et al., 2000]
- Cellule di carcinoma del colon [Prasad et al., 2003]
- Cellule di melanoma [Malafa et al., 2002]
- Cellule di neuroblastoma [Prasad et al., 2003]
- Cellule di linfoma [Sarna et al., 2000]
- Potenzia l’azione antitumorale di diversi chemioterapici come l’adriamicina, il cisplatino e il tamoxifene [Ripoll et al., 1986; Prasad et al., 1994]
- Protegge le cellule del midollo dagli effetti letali della doxorubicina [Fariss et al., 1994].
- Potenzia l’effetto antitumorale di agenti chemioterapici, proteggendo le cellule sane dagli effetti tossici [Prasad et al., 2003]
- Attività antiangiogenetica [Shklar et al., 1996; Tang et al., 2001; Neuzil et al., 2002; Inokuchi et al., 2003; Miyazawa et al., 2004].
MELATONINA
- Azione antiaggregante piastrinica [Di Bella et al., 1969; Di Bella et al., 1979; Di Bella et al., 1980]
- Diminuisce la trascrizione del recettore dell’estrogeno, blocca l’azione mitogena della prolattina e l’effetto blastico indotto dal fattore di crescita epidermico (EGF) [Bartsch et al., 2001]
- Concorre alla sintesi della NO-sintasi, potenziandone la complessa attività anche antitumorale, probabilmente in sinergismo con Ca-modulina, tirosinkinasi, TNF. In questa serie di reazioni che portano sia alla produzione di NO, che alle poliamine, la MLT può esercitare un ruolo fondamentale [Di Bella 1998].
- Modula l’attività ipofiso-gonadica, immunitaria, e l’ azione “scavenger” antitumorale [Di Bella 1998].
- Interagisce con modalità, tempi e meccanismi molteplici con la biologia neoplastica [Di Bella 1997].
- Dispone ubiquitariamente gli esteri fosforici dell’AMP, ADP, ATP [Di Bella 1998].
- nell’ambito del DNES (Sistema Neuroendocrino Diffuso) esercita un ruolo insostituibile stimolando l’apparato sistemico, di risposta e controllo, per la protezione dell’organismo, agendo in tutti i sistemi d’organi. [Kvetnoi et al., 2002]
- Rappresenta la molecola chiave del sistema paracrino per la coordinazione locale delle reazioni intercellulari [Kvetnoi et al., 1994].
- In forma fisiologica, omeostatica, l’organismo tende a normalizzare o contenere i processi proliferativi patologici attraverso la MLT [Kvetnoi et al., 1997].
- La produzione di MLT e dei relativi peptidi APUD, in sito, nei carcinomi non endocrini, svolge un ruolo determinante nei meccanismi autocrini di omeostasi antitumorale [Kvetnoi et al., 1986]
- Riduce l’incidenza di noduli alveolari iperplastici, e la presenza della proteina N-ras, nelle lesioni iperplastiche focali, inoltre previene efficacemente anche l’atipia delle cellule epiteliali e gli adenocarcinomi della mammella, in cui riduce anche l’iperplasia del tessuto linfoide [Mediavilla et al., 1999].
- Rappresenta la molecola chiave del sistema paracrino per la coordinazione distrettuale delle relazioni intercellulari [Maestroni et al., 1988].
- Il tasso plasmatico di MLT è inversamente proporzionale all’indice proliferativo dei tumori, immunoistochimicamente determinato attraverso la presenza dell’antigene nucleare delle cellule proliferanti [Bartsc et al., 2001].
- Effetto antagonizzante sulla crescita prolattino-dipendente del carcinoma umano del seno [Lemus-Wilson et al., 1995].
- Effetto inibente in dosi fisiologiche sulla sintesi di DNA in cellule neoplastiche [Cos et al., 1996].
- Esercita la sua funzione antitumorale anche sugli spazi di giunzione intercellulare inducendo la proteina dello spazio di giunzione CX32 [Kojma et al., 1997]
- Attiva a livello intercellulare il processo di polimerizzazione del tubulin. A concentrazioni fisiologiche induce un aumento di microtubuli nelle cellule tumorali [Melendez et al., 1996].
- Aumenta la radiosensibilità ed esercita effetti stabilizzanti sui disordini metabolici che si sviluppano durante il processo oncologico, esercita capacità immunomodulante, attiva la funzione citotossica dei linfociti natural-killer e la produzione di interferone. [Kvetnoi et al., 1986]
- Esercita azione radioprotettiva e dimostra di possedere proprietà radiomodificanti e radiosensibilizzanti [Lissoni et al., 1996] .
- Se somministrata prima della radioterapia, riduce i danni epatici delle radiazioni ionizzanti. La sua azione radioprotettiva viene attuata attraverso l’inattivazione dei radicali liberi prodotti dalle radiazioni ionizzanti [Taysi et al., 2003].
- Protegge le cellule nervose dallo stress ossidativo indotto dal cobalto, dalla neurotossicità, ed aumenta la secrezione di beta amiloide [Olivieri et al., 2003]. ▪ Previene e ritarda la carcinogenesi chimica [Hrushesky et al., 2009].
- Inibisce, nei pazienti neoplastici, simultaneamente e velocemente sia il rilascio di acido grasso dai corpi adiposi, sia l’assorbimento dell’acido grasso da parte dei tumori [Sauer et al., 2001].
- Esercita effetto antiradicalico sinergico a quello della Vit. E, protegge l’intera cellula dallo stress ossidativo con vari mezzi, tra cui il potenziamento di sistemi enzimatici quali la glutatione perossidasi, l’aumento della sintesi del mRNA e conseguentemente la superossidodismutasi. Inibisce la perossidazione lipidica, con effetto sinergico ai retinoidi. Riduce l’incidenza di mutazioni e pertanto le probabilità di cancro [Reiter et al.2000].
- Inibisce l’increzione di fattori mitogeni come la prolattina [Lemus-Wilson et al., 1995].
- Esercita attraverso i recettori mel1 azione antiproliferativa diretta sulle cellule di cancro umano alla prostata LNCaP androgeno sensibili [Xi et al., 2000].
- Regola diversi messaggeri secondari: cAMP, cGMP, diacilglicerolo, inositolo, ac. Arachidonico e la concentrazione intracellulare di Ca2+. Regola anche i fattori di trascrizione, cioè la fosforilazione della proteina legante, elemento che risponde al cAMP e l’espressione del c-Fos. Esercita meccanismi inibenti l’adenilciclasi, e modulante il metabolismo del fosfolipide e [Ca2+O] [Vanecek 1998].
- Nei talassemici migliora la sintesi di Hb e ne rallenta la degradazione, aumentando anche la resistenza globulare [Di Bella 1998, Di Bella, 1980].
- Effetto antiproliferativo sinergico della MLT e Vit. D3 con capacità delle due molecole di inibire con modaltà dose-dipendente la proliferazione cellulare esprimendo un reciproco e altamente significativo potenziamento anche nell’aumento dell’espressione di TGF-beta che concorre al blocco proliferativo [Bizzarri et al., 2003].
- Mobilizza AR (recettore dell’androgeno) dal cariosol al citosol e ne limita l’espressione, limitando così le risposte epiteliali all’androgeno [Rimler et al., 2002].
- Agisce da agente oncostatico cronobiologico in grado di controllare la proliferazione cellulare e attivare l’apoptosi [Blask et al., 2002].
- Inibisce i tumori estrogeno-dipendenti riducendo l’espressione e la trascrizione del recettore dell’estrogeno, la pervietà dei canali ionici delle membrane cellulari al calcio, l’attività delle proteinchinasi, l’architettura e la funzionalità citoscheletrica, la veicolazione, la metabolizzazione e l’utilizzazione dell’acido linoleico e altri acidi grassi da parte delle cellule neoplastiche. Sopprime EGFR (recettore del fattore di crescita epidermico) [Blask et al., 2002].
- Inibisce reversibilmente la proliferazione neoplastica, mentre aumenta l’espressione delle proteine P53 e P21 WAF 1, regola il ciclo cellulare e l’incidenza di metastasi mediante espressione delle proteine di adesione cellulare E-caderina e di beta-1 integrina; riduce inoltre l’espressione di ER e la risposta del DNA al complesso ER [Pawlikowski et al. 1999-2002].
- Inibisce la diffusione metastatica delle cellule tumorali. L’azione si realizza attraverso una ridotta attrazione per la fibronectina [Mediavilla et al.,1999].
- Incrementa sensibilmente nei pazienti neoplastici l’aspettativa di vita migliorandone la qualità [ Lissoni et al., 1999]
- Contiene decisamente i processi che portano alla cachessia neoplastica [Lissoni et al., 1999]
- Riduce la tossicità della chemioterapia [ Lissoni et al., 1999]
- La quantità di cellule neoplastiche che i tessuti possono elaborare, e liberare, è condizionata dalla funzione inibente antitumorale della MLT e dalla sua concentrazione nel sangue e nei tessuti [Di Bella 1997].
Somatostatina e analoghi
- Aumenta l’espressione della topo isomerasi, inibendo il ciclo proliferativo di cellule neoplastiche [Brevini et al., 2001].
- Inibizione dei percorsi non ossidativi del pentosio fosfato [Boros et al., 1998].
- Inibizione del riciclo del carbonio tramite il PC del 5,7%, con aumento al 19,8% in combinazione con l’ossitiamina [Boros et al .1998].
- Regolazione dei canali ionici, inibizione dell’adenilciclasi, della chinasi e della serina/treonina fosfatasi e tiroxina fosfatasi [Bousquet et al., 2001].
- Forte aumento dell’attività della adenilato ciclasi [Giannetti et al., 2000].
- Inibizione della sintesi del DNA [Charland et al., 2001].
- Effetto antiproliferativo attraverso la soppressione della riduzione di p27 [Baradari et al., 2006]
- Induzione dell’espressione di p21Cip, inibizione del percorso del fosfoditilinositolo chinasi -3, e una maggior espressione di p21Cip e p27Kip1, che porta alla repressione della fosforilazione del pRb e della complessa attività di cyclin E-cdk2 [Charland et al., 2001].
- Inibizione dell’incorporazione della 3H-timidina nel DNA delle cellule tumorali [Yano et al., 2000; Feind et al., Damge et al., 1998].
- Riduzione significativa di IGF-1 [Ingle et al., 1999].
- Inibizione, con modalità dose-dipendente della fosforilazione tirosinica, da parte di EGFR (attivato dal EGF) [Mischima et al., 1999].
- Induzione della traslocazione del PTP1C intracellulare, alle membrane di cellule neoplastiche [Srikant et al., 1996].
- Induzione mediata dagli SSTR, dell’attività della tirosin-fosfatasi di membrana (PTP), implicata nella segnalazione antiproliferativa per la sua capacità di defosforilare e inattivare le chinasi del recettore del fattore di crescita [Srikant et al., 1996].
- Inibizione PTPase e PTP1C, e dell’attività di tirosina chinasi della membrana e di p 42MAP chinasi [Douziech et al., 1999].
- Riduzione nelle cellule tumorali dei recettori del fattore di crescita epidermico EGFR [Szepeshazi et al., 1999].
- Effetto positivo e stimolante sulle cellule di Kupfer, con meccanismo antitumorale, potenziato da una decisa inibizione della perossidazione lipidica epatica [Kouroumanlis et al., 2001].
- Effetto apoptotico con condensazione nucleare della cromatina e frammentazione, restringimento cellulare, e formazione di corpi apoptotici, con una correlazione direttamente proporzionale, dose-dipendente, tra concentrazione di somatostatina e tasso apoptotico [Che et al., 2009].
- Inibizione della fase S del ciclo cellulare con induzione dell’apoptosi dose-dipendente, aumento della perossidazione lipidica intrametastatica, con perdita dell’integrità delle cellule tumorali [Raderer et al.2000].
- Abbattimento della concentrazione plasmatica di fattori di crescita tumorale come l’IGF-1 e l’EGF con netta diminuzione della percentuale della fase S statisticamente significativa [Cascinu et al., 1997].
- Aumento dell’attività del gene soppressore p53, con la capacità inibente sulle linee di tumori, del tutto indipendentemente dallo stato del loro p53 [Szepeshasi et al., 2002].
- Potenziamento dell’attività dei chemioterapici nei tumori [Tesei et al., 2000].
- Inibizione dell’attività di chinasi della proteina mitogeno attivata MAB [Cattaneo et al., 2000].
- Intensa attività fosfatasica [Cattaneo et al., 1996].
- Soppressione dell’attivazione del Ras indotto da PDGF [Cattaneo et al., 1999].
- Induzione non solo all’apoptosi ma alla CA (aberrazione cromosomica), cioè rottura cromosomica con deciso effetto antiblastico [Tompa et al., 2000].
- Induzione della migrazione delle cellule della AML mediante l’attivazione di SSTR-2 ed attrazione sulle normali cellule progenitrici emopoietiche, proprietà chemiotattiche, con implicazioni nella distribuzione delle cellule AML nel corpo, con applicazioni cliniche nella leucemia mieloide acuta [Oomen et al., 2001].
- Attivazione delle fosfatasi della tiroxina, della proteina SHP2 e inibizione delle chinasi della proteina mitogeno-attivata [Held Feind et al., 2000].
- Inibizione in maniera significativa, dose-dipendente, della proliferazione di cellule leucemiche con riduzione dell’espressione del gene c-fos [Ishihara et al., 1999].
- Induzione di una forte espressione della proteina bcl-2 con relativo effetto apoptotico [Zalatnai et al., 1999].
- Diminuzione delle cellule in fase S e dell’indice proliferativo dose dipendente [Raderer et al., 2000].
- Diminuzione dei livelli sierici di AFP negli epatocarcinomi [Raderer et al., 2000].
- Defosforilazione delle chinasi della proteina mitogeno attivata ERK 1-2 [Held Field et al., 2001].
- Riduzione dell’espressione di EGF stimolata dal complesso AP1 a livello trascrizionale e traslazionale [Held Field et al., 2001].
- Effetto proapoptotico e antiproliferativo sinergico con MLT [Melen-Mucha et al., 1998].
Vit. D3 e analoghi
- Induzione di differenziazione, apoptosi, blocco proliferativo di progressione alla fase S per l’apparire della forma ipofosforilata della proteina del retinoblastoma [pRb] inibente crescita e attività di modulazione delle chinasi ciclino-dipendenti [cdk] 2-4-6. [Jensen et al., 2001].
- La D3 impedisce l’attivazione della ciclina D1cdk-4, e la perdita della ciclina D3, che insieme portano alla perdita dei fattori di trascrizione di E2F, inibendo l’espressione della proteina A della ciclina. Insieme a una rapido decremento dell’oncoproteina c-Myc in risposta alla D3, questi risultati dimostrano che D3 intervenendo su regolatori chiave della transizione G1-S, blocca la proliferazione [ Jensen et al., 2001].
- Attività pro-differenziante della D3, che si realizza non solo interagendo col recettore, ma anche con meccanismi extrarecettoriali mediati dalla membrana [Marcinkowska 2001].
- Inibizione sia dell’espressione di PTHR nell’osso diminuendone la trascrizione mediante P2, che della trascrizione del gene di PTHrP. Il dato è clinicamente rilevante, per evitare i gravi danni prodotti dall’ipercalcemia indotta da sovraproduzione di PTHrP nelle cellule tumorali [Goltzman 2001].
- Inibizione dell’angiogenesi, dello sviluppo e crescita indotti dal fattore di crescita endoteliale vascolare VEGF delle cellule endoteliali, in modo dipendente dalla dose, inibizione della formazione di cellule endoteliali allungate all’interno dei gel di collageno3D, con regressione dovuta all’induzione all’apoptosi [Mantell et al.,2000] .
- Attivazione di un recettore nucleare specifico per inibire la proliferazione e promuovere la differenziazione di numerosi tipi di cellule tumorali, inibizione inoltre dell’adesione e migrazione delle cellule dalla membrana basale, dovuta ad una diminuzione dell’espressione degli integrins alpha-6 e beta-4, che sono recettori della laminina associati ad una maggiore migrazione ed invasione delle cellule di cancro alla prostata in vivo [Sung et al.,2000].
- Induzione dell’espressione di mRNA della proteina di BRCA1, e dell’attivazione trascrizionale da parte del promotore di BRCA1. Infatti la sensibilità agli effetti antiproliferativi della Vit. D3, è intimamente collegata alla 16capacità di modulare la proteina di BRCA1 mediante attivazione trascrizionale dei fattori indotti da VDR [Campbell et al., 2000]
- L’attivazione del VDR, oltre all’effetto antiproliferativo, aumenta l’espressione della proteina legante il fattore di crescita simil-insulinico IGF [Chokkalingam et al 2001].
- Incrementa l’espressione della proteina 3 legante l’IGF (IGFBP3), la cui presenza è indispensabile per attivare l’effetto antiproliferativo della D3. Sia la D3 che IGFBP3 attivano la proteina inibitoria della chinasi ciclin-dipendente p21/WAF 1, che media il loro effetto antiproliferativo [Boyle et al., 2001].
- Inibisce la segnalazione del fattore di crescita dei cheratinociti e induce apoptosi nelle cellule di cancro umano della prostata, induce la diminuzione dell’espressione basale di bcl 2, con relativo effetto [Crescioli et al., 2006 ].
- Riduce l’effetto di stimolazione della crescita del DHT, e incrementa l’espressione di VDR [Ahnonen et al., 2000].
- Influenza la comunicazione intercellulare degli spazi di giunzione (GJIC) durante la carcinogenesi aumenta la funzione di GJIC dei HRPTC [Fujioka et al., 2000].
- Induce la maturazione fenotipica delle cellule tumorali in cellule funzionalmente mature, differenziate, fisiologicamente normali, attiva parallelamente un’inibizione della proliferazione cellulare neoplastica potenziando l’effetto antiproliferativo dell’ac. Trans-retinoico [Barroga et al., 2000].
- Inibisce l’invasività della matrice extracellulare e le metastasi attraverso il blocco della degradazione delle barriere della matrice extracellulare (ECM) da parte delle cellule tumorali mediante la collagenolisi [Yudoh et al., 1999].
- Inibisce irreversibilmente la crescita e blocca in G0-G1 la mitosi cellulare neoplastica con forte inibizione della clono-proliferazione e invasività [Hisatake et al., 2001].
- Determina un accumulo di cellule in G0-G1, e la successiva apoptosi [Blutt et al., 2000].
- Inibisce l’angiogenesi tumorale, oltre ad esercitare effetti antiproliferativi, prodifferenzianti, proapoptotici, fortemente potenziati dal sinergismo con i retinoidi [Majewski et al., 1994].
- Blocca in fase G1 il ciclo cellulare neoplastico, impedendo la proliferazione cellulare, e abbattendo le concentrazioni di cyclin C e D1, noti attivatori della riproduzione cellulare [Verlinden et al., 2000].
- Promuove selettivamente l’espressione delle molecole di adesione ICAM-3, in modo dipendente dal tempo e dalla dose [Babina et al., 2000].
- Inibisce l’espressione della proteina anti-apoptotica Bcl-2, favorendo conseguentemente l’apoptosi [Larsen et al., 2001].
- Induce all’apoptosi, attraverso il coinvolgimento della fosfolipasi A2 citosolica, inducendo frammentazione di DNA e perdita di vitalità delle cellule neoplastiche [Pirianov et al., 1999].
- Rafforza la risposta delle cellule tumorali al TNF-alfa [Pirianov et al., 1999 ].
- Disattiva l’effetto antiapoptotico dell’inibitore delle caspasi ad ampio spettro Z VADFMK [Pirianov et al., 2001].
- Attiva un’altra via apoptotica caspasi indipendente, mediata dal coinvolgimento della cerammide e fosfolipasi A-2 (cPLA2) [Pirianov et al., 1999].
- Esercita attività antiproliferativa attraverso l’induzione del gene l’amphiregulin e l’aumento del suo mRNA. Inibisce così l’EGF, su cui agisce l’amphiregulin [Akutsu et al., 2001].
- Esprime attività antimitotica direttamente proporzionale alla concentrazione di 1 alpha OH-ase e inversamente a quella di 24OH-ase [Bareis et al., 2001].
- Induce E-caderina e altre molecole di adesione, con effetto proapoptotico [Palmer et al., 2001].
- Inibisce significativamente la perossidazione epatica dei lipidi citosolici e protegge le membrane cellulari dai radicali liberi. Esercita un effetto protettivo massimo sulla normale architettura cellulare degli epatociti e mantiene la concentrazione del citocromo epatico P 450 a livello fisiologico [Basak et al., 2001].
- Esercita, anche mediante meccanismi non recettoriali, una potente azione antiproliferativa e prodifferenziante [Consolini et al., 2001].
- Esercita effetti antiproliferativi sinergicamente potenziati dall’acido retinoico con abbattimento dei livelli della proteina c-myc [Stio et al., 2001].
- Induce una maggior espressione nucleare della proteina dell’inibitore della chinasi dipendente dalla ciclina P27 (kip 1 ) [Liu et al., 2002].
- Induce un’elevata espressione di P21 e P27, regolatori del ciclo cellulare. [Johnson CS 2006].
- Incrementa l’espressione di p27, codificatore degli inibitori delle chinasi cyclin-dipendenti e di gadd4alfa, gene dell’arresto della crescita e dei danni al DNA [Prudencio et al., 2001].
- Blocca l’espressione del recettore EGF attraverso l’inibizione della sua fosforilazione, con defosforilazione dei polipeptidi 17 e 66-kDa, recettori di EGF [Lee et al., 2001].
- Riduce la presenza di cellule CD34(+) con effetto immunostimolante [Lathers et al., 2001].
- Promuove il clivaggio della molecola che segnala la promozione della sopravvivenza e della crescita attivata dal mitogeno (protein Kinase) con meccanismo caspasi dipendente. L’apoptosi avviene attraverso il clivaggio selettivo caspasi dipendente del MEK-1 ed è mediata dal p38 MAPK [Mc Guire et al., 2001].
- Abbatte le concentrazioni di cyclin C e D1 noti attivatori della riproduzione cellulare [Verlinden et al., 2000].
- Promuove l’espressione di molecole di adesione ICAM 3, agisce sui mastocidi di leucemia [Babina et al., 2000].
Vitamina C
- L’acido ascorbico è uno dei più importanti agenti riducenti presenti nei tessuti viventi, è un forte agente anti-ossidante, che reagisce direttamente con atomi di ossigeno singoli, idrossidi e radicali superossidi [Sauberlich 1994].
- I linfociti umani normali hanno la capacità di concentrare intracellularmente la Vit.C, che aiuta a proteggere tali cellule dai danni ossidativi [Levine et al. 1996; Ozturk et al. 2001].
- Previene i danni cellulari indotti da prodotti ossidativi, inclusi i radicali liberi [Padh 1991]
- E’ documentata una relazione inversa statisticamente significativa tra la quantità di Vit.C, caroteni, verdure ed agrumi consumati e l’incidenza di linfoma non-Hodgkin [Ward et al. 1994].
- Può avere un ruolo preventivo e terapeutico nel cancro [Bendich and Langseth 1995].
- Inibisce e gli effetti carcinogenici prodotti da sostanze mutagene [Aidoo et al. 1994; Lee et al. 2002].
- Preserva l’integrità del tessuto connettivo inn funzione antiblastica [Bendich and Langseth 1995].
- Esercita attività angiostatica sulla proliferazione delle cellule endoteliali [Ashino et al. 2003].
- Cellule-T di linfoma NH, sono sensibili alla Vit.C. Concentrazioni minori di 50 micromol/l uccidono le cellule nel giro di poche ore [Helgestad et al. 1990].
- Linee cellulari di tumori linfoblastici, sono inibite dalla Vit.C [Kao et al. 1993].
- Esercita attività antineoplastica con diversi meccanismi d’azione [Cameron et al., 1979; Head 1998].
- Esercita attività antimetastatica mediante la sintesi di collagene [Pinnel et al. 1987; Peterkofsky 1991]
- Esercita attività antimetastatica attraverso linibizione della ialuronidasi [Cameron et al., 1973].
- Esercita attività antimetastatica diminuendo la permeabilità di cellule endoteliali alle popolazioni cellulari neoplastiche [Utoguchi et al. 1995].
- Migliora il performance status nei pazienti neoplastici [Head 1998].
- Incrementa la sopravvivenza dei pazienti neoplastici terminali [Cameron et al., 1974; Cameron et al., 1976; Cameron et al., 1978; Cameron 1991].
- Potenzia l’efficacia di farmaci antineoplastici in cellule di linfoma [Michel et al. 2003; Nagy et al. 2003; Lee et al., 1994; Prasad et al. 1994; Kurbacher et al. 1996; Nagy et al., 2003; Prasad et al. 1992; Sarna et al.,1993].
- Riduce della tossicità di agenti chemioterapici come l’adriamicina [Fujita et al. 1982; Shimpo et al. 1991]
Bromocriptina e/o Cabergolina
Inibitori della Prolattina , di cui sono documentate le seguenti attività mitogeniche :
Inibitori della Prolattina , di cui sono documentate le seguenti attività mitogeniche :
- forte induzione mitogena [Ben-Jonathan et al. 2002].
- aumento dell’aggressività dei carcinomi colonrettale [Bhatavdekar et al. 1994; Bhatavdekar et al. 1995]
- induzione della proliferazione di diverse linee di cancro del seno umano [Vonderhaar 1998; Vonderhaar 1999]
- Stimola la proliferazione di cellule di cancro della prostata [Janssen et al. 1996]
- Attiva la proliferazione di cellule di leucemia acuta mieloide [Nishiguchi et al. 1993]
- Regola positivamente la proliferazione di cellule di leucemia acuta linfoide [Matera et al. 1997]
- Incrementa la proliferazione di linfociti B maligni [Walker et al. 1994]
- Eleva l’indice proliferativo di cellule di linfoma [Gout et al.1980; Yu-Lee 1990]
- Nelle cellule maligne del sistema immunitario, inibisce il processo apoptotico [Krumenacker et al. 1998; Buckley and Buckley 2000].
- Il recettore della prolattina è espresso dalla maggior parte delle cellule del sistema immunitario, [O'Neal et al.1991; Dardenne et al. 1994; Matera et al. 2000]
- Favorisce il processo di epatocarcinogenesi [Buckley et al. 1988]
- I leiomiomi producono più prolattina rispetto al normale miometrio, esercitando atttreverso la prolattina prodotta localmente un’azione mitogenica [Nowak et al. 1993].
- Azione autocrina/paracrina della prolattina nelle cellule emopoietiche [Matera 1996; Ben-Jonathan et al. 2002].
- Il recettore della prolattina è espresso dalla maggior parte delle cellule maligne del sistema immunitario, [O'Neal et al. 1991; Dardenne et al. 1994; Matera et al. 2000].
- Le cellule emopoietiche maligne possono produrre prolattina. È stato riportato che cellule leucemiche mieloidi, così come mieloblasti isolati da pazienti con leucemia acuta producono la prolattina [Kooijman et al. 2000].
- Diverse linee cellulari di linfoma non-Hodgkin producono la prolattina [Matera et al. 2000].
- La linea cellulare di linfoma di ratto, Nb2, dipende dalla prolattina per la crescita. [Davis et al., 1988; LaVoie et al.,1995; Ganguli et al. 1996; Camarillo et al. 1997; Camarillo et al., 1998; Krumenacker et al. 1998; Al-Sakkaf et al. 2000; Yu et al., 2000]. 20
- Partecipa allo sviluppo e/o alla progressione di neoplasie ematologiche [Hooghe et al. 1998].
Obiettivi del MDB
il MDB persegue 3 obiettivi essenziali:
a) La difesa dall’aggressione neoplastica
b) L’inibizione della proliferazione neoplastica
c) Il contrasto della spiccata tendenza mutagena del fenotipo neoplastico.
a) La difesa dall’aggressione neoplastica
b) L’inibizione della proliferazione neoplastica
c) Il contrasto della spiccata tendenza mutagena del fenotipo neoplastico.
DIFESA
Il MDB asseconda ed esalta le reazioni vitali e l’omeostasi antitumorale per metterle in condizione di contrapporsi alla insorgenza e progressione neoplastica .
Il tumore è deviazione dalla vita normale, per cui occorre riportare le reazioni deviate verso la norma, attraverso il potenziamento di tutti quei mezzi che la Fisiologia considera essenziali per la vita [Di Bella et al. , 1969; Di Bella et al. ,1971; Di Bella et al., 1974; Di Bella et al.,1976; Di Bella et al., 1977; Di Bella et al., 1979; Di Bella et al., 1980; Di Bella et al., 1981; Di Bella et al., 1984; Di Bella et al., 1985; Di Bella et al., 1986; Di Bella et al., 1987; Di Bella et al., 1988; Di Bella et al., 1994; Di Bella 1997; Di Bella et al., 1998; Di Bella et al., 2002; Di Bella et al., 2006].
Il MDB persegue questo obiettivo attraverso innovative formulazioni e criteri d’impiego della MLT (complessata con Adenosina e Glicina), di retinoidi solubilizzati in Vit E, oltre a Vitamine C, D3, e componenti della ECM. Inserendo componenti apolari come il Betacarotene e la vit.E tra i fosfolipidi di una membrana cellulare, la si stabilizza preservandola da danni ossidativi e dai radicali liberi [Shklar et al., 1996; Israel et al., 2000; Khuri et al., 2001; Di Bella , 2005; Dong et al., 2008; Lubin et al., 2008; Nesaretnam et al., 2008; Watters et al., 2009].
Sia nelle situazioni che predispongono al tumore, che nel corso della malattia neoplastica, possono essere sovvertiti struttura e potenziali della membrana cellulare e conseguentemente, l’espressione e le funzionalità recettoriali, mediante l’esasperazione dei processi ossidativi e il conseguente picco nella produzione diradicali liberi. Le dosi, previste dal MDB, di retinoidi e vit. E, permettono di conseguire sia un effetto preventivo, che terapeutico, azzerando le possibilità che i radicali liberi possano provocare danni, [Odeleye et al., 1992; Launoy et al., 1998;Shimizu et al., 2004; Di Bella, 2005; Elangovan et al., 2008; Neuzil et al., 2002; Frei et al., 2008].
L’ obiettivo di ottimizzare le reazioni vitali difendendole dall’aggressione neoplastica si realizza con:
Retinoidi [Kapil et al., 1993; Lotan et al., 1997; Muller et al., 1997; Roth et al., 1999; Khuri et al., 2001; Hashimoto et al., 2003; Di Bella G. 2005; Barroga et al.,2000; Dong et al., 2008; Bonofiglio et al., 2009]
Vit. E [Israel et al., 2000; Jatoi et al., 2002; Neuzil et al., 2002; Di Bella, 2005; Lubin et al., 2008; Elangovan et al., 2008; Nesaretnam et al., 2008];
Vit. D3 [Meggouh et al., 1990; Di Bella, 2005; Giovannucci et al., 2006; Amir et al.,2009; Chiang et al., 2009; Chung et al., 2009; Reicharth et al., 2009; Schwartz et al., 2009; Fernandez et al., 2009; Goodwin et al., 2009]
Vit. C [Cameron et al., 1979; Murata et al., 1982; Di Bella, 2005; Frei et al., 2008; Ha et al., 2009;]
MLT [Di Bella et al. 1971; Di Bella et al., 1974; Di Bella et al.,1976; Di Bella et al.,1977; Di Bella et al., 1979; Di Bella et al., 1980; Di Bella et al., 1981; Di Bella et al., 1984; Kvetnoĭ et al., 1986; Di Bella et al., 1988; Maestroni et al., 1996; Di Bella et al., 1997; Di Bella et al., 1998; Cos et al., 2000; Bartsch et al., 2001; Di Bella et al.,2002; Di Bella 2005; Di Bella et al., 2006; Joo et al., 2009; Di Bella et al., 2009;Grant et al., 2009; Di Bella et al., 2009; Di Bella et al., 2009]
Componenti della Matrice Extracellulare (ECM) [Batra et al., 1997; Kidd et al., 2000; Mikami et al., 2001; Di Bella 2005; Asimakopoulou et al., 2008; Yamada et al., 2008].
I Retinoidi e la Melatonina hanno la capacità di preservare ed esaltare il trofismo, la vitalità e l’efficienza delle cellule sane, nello stesso momento in cui deprimono la progressione, la vitalità e la spiccata attitudine mutagena del fenotipo neoplastico [DiBella et al., 1979; Di Bella et al., 1997; Onogi et al., 1998; Mediavilla et al., 1999; Bartsch et al., 1999; Wang et al., 1999; Khuri et al., 2001; Di Bella 2005; Di Bella et al., 2006; Garcia-Santos et al., 2006; Bogos et al., 2008; Martín-Renedo et al., 2008; Yap et al., 2008; Watters et al., 2009; Williams et al., 2009; Wu et al., 2009; Ginestier et al., 2009; Di Bella et al., 2009;Yin et al., 2009; Kim et al., 2009; Di Bella et al., 2009].
Questa apparente contraddizione deriva dal fatto che i retinoidi sono i più potenti attivatori, non ormonali, unicamente della crescita ordinata, funzionale e finalizzata all’equilibrio biologico ottimale, mentre allo stesso tempo inibiscono decisamente l’afinalistica e disordinata crescita neoplastica, avviando la cellula tumorale all’apoptosi.
Le vitamine sono catalizzatori fisiologici fra energia e materia.
Ogni cambiamento della materia vivente non può prescindere da un adeguamento dello stato energetico. Solo minime variazioni quantitative di produzione,
assorbimento, cioè elaborazione del terreno biologico e del suo corrispettivo energetico, sono compatibili con la vita, e quindi le reazioni devono procedere per passaggi graduali di entità minima materiali-energetiche, reciprocamente compensati nel tempo. Queste reazioni realizzano, con estrema gradualità, la produzione e l’assorbimento di energia e materia con equivalenza materiale/energetica. Questo continuo divenire, per le eccezionali finalità cui tende, deve essere gradualmente modulato e finemente regolato, e nelle sue linee essenziali sarebbe impossibile senza le vitamine, il cui fine è il condizionamento e la regolazione dell’equilibrio materia/energia su cui poggia la vita [Di Bella 2005].
La piena conoscenza delle vitamine equivale alla conoscenza dei più fini equilibri e dei rapporti energia/materia e di tutti i riflessi sull’attività vitale. La conoscenza della composizione chimica, della formazione, della localizzazione all’interno della cellula,del momento del loro intervento, della regolazione e dell’entità della loro attività, consente di cogliere l’essenza della vita fisiologica e di correggere le sue deviazioni patologiche. Perciò, dal suo ruolo originario biochimico-vitale, la vitaminologia è elevata, nel MDB, a quello terapeutico razionale, essenziale, sia nella prevenzione, che nella cura di varie patologie. Pertanto la conoscenza approfondita dei meccanismi regolatori della vita normale, fisiologica, consente la predisposizione di contromisure efficaci per evitare deviazioni degenerative o neoplastiche [Di Bella 2005].
Il tumore è deviazione dalla vita normale, per cui occorre riportare le reazioni deviate verso la norma, attraverso il potenziamento di tutti quei mezzi che la Fisiologia considera essenziali per la vita [Di Bella et al. , 1969; Di Bella et al. ,1971; Di Bella et al., 1974; Di Bella et al.,1976; Di Bella et al., 1977; Di Bella et al., 1979; Di Bella et al., 1980; Di Bella et al., 1981; Di Bella et al., 1984; Di Bella et al., 1985; Di Bella et al., 1986; Di Bella et al., 1987; Di Bella et al., 1988; Di Bella et al., 1994; Di Bella 1997; Di Bella et al., 1998; Di Bella et al., 2002; Di Bella et al., 2006].
Il MDB persegue questo obiettivo attraverso innovative formulazioni e criteri d’impiego della MLT (complessata con Adenosina e Glicina), di retinoidi solubilizzati in Vit E, oltre a Vitamine C, D3, e componenti della ECM. Inserendo componenti apolari come il Betacarotene e la vit.E tra i fosfolipidi di una membrana cellulare, la si stabilizza preservandola da danni ossidativi e dai radicali liberi [Shklar et al., 1996; Israel et al., 2000; Khuri et al., 2001; Di Bella , 2005; Dong et al., 2008; Lubin et al., 2008; Nesaretnam et al., 2008; Watters et al., 2009].
Sia nelle situazioni che predispongono al tumore, che nel corso della malattia neoplastica, possono essere sovvertiti struttura e potenziali della membrana cellulare e conseguentemente, l’espressione e le funzionalità recettoriali, mediante l’esasperazione dei processi ossidativi e il conseguente picco nella produzione diradicali liberi. Le dosi, previste dal MDB, di retinoidi e vit. E, permettono di conseguire sia un effetto preventivo, che terapeutico, azzerando le possibilità che i radicali liberi possano provocare danni, [Odeleye et al., 1992; Launoy et al., 1998;Shimizu et al., 2004; Di Bella, 2005; Elangovan et al., 2008; Neuzil et al., 2002; Frei et al., 2008].
L’ obiettivo di ottimizzare le reazioni vitali difendendole dall’aggressione neoplastica si realizza con:
Retinoidi [Kapil et al., 1993; Lotan et al., 1997; Muller et al., 1997; Roth et al., 1999; Khuri et al., 2001; Hashimoto et al., 2003; Di Bella G. 2005; Barroga et al.,2000; Dong et al., 2008; Bonofiglio et al., 2009]
Vit. E [Israel et al., 2000; Jatoi et al., 2002; Neuzil et al., 2002; Di Bella, 2005; Lubin et al., 2008; Elangovan et al., 2008; Nesaretnam et al., 2008];
Vit. D3 [Meggouh et al., 1990; Di Bella, 2005; Giovannucci et al., 2006; Amir et al.,2009; Chiang et al., 2009; Chung et al., 2009; Reicharth et al., 2009; Schwartz et al., 2009; Fernandez et al., 2009; Goodwin et al., 2009]
Vit. C [Cameron et al., 1979; Murata et al., 1982; Di Bella, 2005; Frei et al., 2008; Ha et al., 2009;]
MLT [Di Bella et al. 1971; Di Bella et al., 1974; Di Bella et al.,1976; Di Bella et al.,1977; Di Bella et al., 1979; Di Bella et al., 1980; Di Bella et al., 1981; Di Bella et al., 1984; Kvetnoĭ et al., 1986; Di Bella et al., 1988; Maestroni et al., 1996; Di Bella et al., 1997; Di Bella et al., 1998; Cos et al., 2000; Bartsch et al., 2001; Di Bella et al.,2002; Di Bella 2005; Di Bella et al., 2006; Joo et al., 2009; Di Bella et al., 2009;Grant et al., 2009; Di Bella et al., 2009; Di Bella et al., 2009]
Componenti della Matrice Extracellulare (ECM) [Batra et al., 1997; Kidd et al., 2000; Mikami et al., 2001; Di Bella 2005; Asimakopoulou et al., 2008; Yamada et al., 2008].
I Retinoidi e la Melatonina hanno la capacità di preservare ed esaltare il trofismo, la vitalità e l’efficienza delle cellule sane, nello stesso momento in cui deprimono la progressione, la vitalità e la spiccata attitudine mutagena del fenotipo neoplastico [DiBella et al., 1979; Di Bella et al., 1997; Onogi et al., 1998; Mediavilla et al., 1999; Bartsch et al., 1999; Wang et al., 1999; Khuri et al., 2001; Di Bella 2005; Di Bella et al., 2006; Garcia-Santos et al., 2006; Bogos et al., 2008; Martín-Renedo et al., 2008; Yap et al., 2008; Watters et al., 2009; Williams et al., 2009; Wu et al., 2009; Ginestier et al., 2009; Di Bella et al., 2009;Yin et al., 2009; Kim et al., 2009; Di Bella et al., 2009].
Questa apparente contraddizione deriva dal fatto che i retinoidi sono i più potenti attivatori, non ormonali, unicamente della crescita ordinata, funzionale e finalizzata all’equilibrio biologico ottimale, mentre allo stesso tempo inibiscono decisamente l’afinalistica e disordinata crescita neoplastica, avviando la cellula tumorale all’apoptosi.
Le vitamine sono catalizzatori fisiologici fra energia e materia.
Ogni cambiamento della materia vivente non può prescindere da un adeguamento dello stato energetico. Solo minime variazioni quantitative di produzione,
assorbimento, cioè elaborazione del terreno biologico e del suo corrispettivo energetico, sono compatibili con la vita, e quindi le reazioni devono procedere per passaggi graduali di entità minima materiali-energetiche, reciprocamente compensati nel tempo. Queste reazioni realizzano, con estrema gradualità, la produzione e l’assorbimento di energia e materia con equivalenza materiale/energetica. Questo continuo divenire, per le eccezionali finalità cui tende, deve essere gradualmente modulato e finemente regolato, e nelle sue linee essenziali sarebbe impossibile senza le vitamine, il cui fine è il condizionamento e la regolazione dell’equilibrio materia/energia su cui poggia la vita [Di Bella 2005].
La piena conoscenza delle vitamine equivale alla conoscenza dei più fini equilibri e dei rapporti energia/materia e di tutti i riflessi sull’attività vitale. La conoscenza della composizione chimica, della formazione, della localizzazione all’interno della cellula,del momento del loro intervento, della regolazione e dell’entità della loro attività, consente di cogliere l’essenza della vita fisiologica e di correggere le sue deviazioni patologiche. Perciò, dal suo ruolo originario biochimico-vitale, la vitaminologia è elevata, nel MDB, a quello terapeutico razionale, essenziale, sia nella prevenzione, che nella cura di varie patologie. Pertanto la conoscenza approfondita dei meccanismi regolatori della vita normale, fisiologica, consente la predisposizione di contromisure efficaci per evitare deviazioni degenerative o neoplastiche [Di Bella 2005].
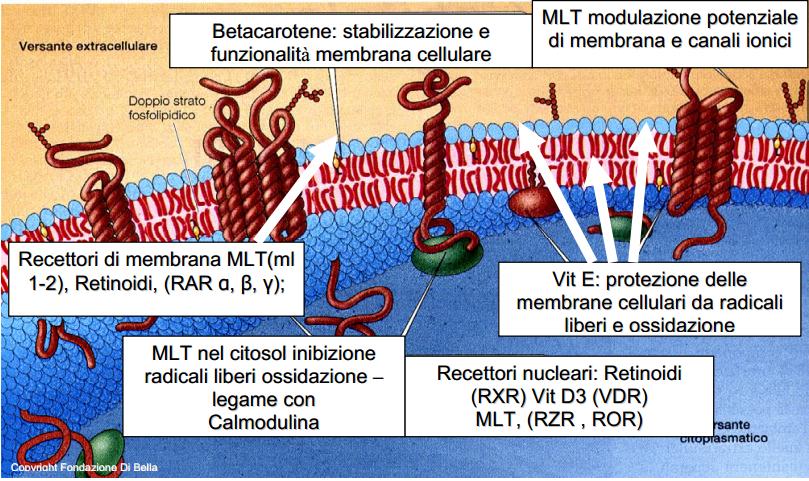
| La membrana cellulare (in azzurro, contenente lo strato fosfolipidico in rosso ) è una difesa, un filtro vitale attraverso cui tutto transita, dall’interno della cellula all’esterno, dove vengono recepiti e analizzati gli stimoli e i condizionamenti, dall’esterno all’interno e viceversa, in cui avviene la comunicazione, vengono emessi e ricevuti impulsi e segnali. Ottimizzarla, renderla efficiente, vuol dire rendere la cellula capace di difendersi in condizioni ottimali, potenziarla: la Vit.E ed il Betacarotene proteggono e stabilizzano la membrana, la MLT ne modula fisiologicamente i potenziali, regolando i canali ionici e tutta la dinamica ed espressione recettoriale. Per comprendere l’enorme valenza dei retinoidi nell’ambito dell’economia biologica, basta considerare che essi forniscono l’alto costo energetico sia della crescita, che dell’ordine fisiologico della crescita stessa, concorrendo all’omeostasi antitumorale. La crescita della sostanza vivente comporta un altissimo dispendio energetico, ma l’ordine fisiologico della crescita comporta un pari, ed ugualmente elevato, fabbisogno di energia. |
INIBIZIONE DELLA PROLIFERAZIONE NEOPLASTICA
L’espressione recettoriale ubiquitaria della Prolattina e del GH [De Souza et al., l974; Di Bella et al., 1979; Di Bella et al., 1981; Di Bella et al., 1997; Hooghe et al., 1998; Di Bella et al., 1998; Ben-Jonathan et al., 2002; Di Bella 2005; Di Bella 2009] rappresenta uno degli aspetti del ruolo mitogeno, diretto e generalizzato, di queste molecole.
La proliferazione cellulare è strettamente dipendente dalla Prolattina [Bonneterre et al., 1990; Di Bella et al., 1997; Di Bella et al., 1998;Tada et al., 1999; Gruszka et al., 2001; Di Bella 2005; Florio et al., 2008; Mouton 2008; Di Bella 2009;] dal GH, massimo fattore di crescita [De Souza et al., l974; Di Bella et al., 1979; Di Bella et al., 1997; Di Bella L et al., 1998; Lincoln et al., 1998; Friend et al., 2000; Barnett et al., 2003; Anthony et al., 2009;] e da molecole mitogene GH dipendenti, da esso positivamente regolate, come EGF, FGF, HGF, IGF1-2, NGF, PDGF, TGF, VEGF [Szepesházi et al., 1999; Murray et al., 2004; Sall et al., 2004; Di Bella 2009; Hagemeister et al., 2008; Di Bella et al., 2009; Taslipinar et al., 2009] oltre che da fattori di crescita prodotti dall’apparato gastrointestinale, come VIP, CCK, G [Kath et al., 2000].
Sia la proliferazione cellulare fisiologica, che quella neoplastica, avvengono per mezzo di queste stesse molecole, che la cellula neoplastica utilizza, però, in rapporto esponenziale rispetto a quella sana. La perdita di differenziazione e la proliferazione incontrollata, anche se in misura diversa, caratterizzano tutte le neoplasie.
L’impiego della somatostatina e analoghi, agendo sulla crescita, denominatore comune a ogni tumore, deve trovare indicazione razionale in ogni neoplasia [Di Bella et al., 1979; Di Bella et al., 1981; Di Bella et al., 1997; Pollak et al., 1997; Di Bella et al., 1998; Pawlikowski et al., 1998; Friend et al., 2000; Lachowicz et al., 2000; Friend et al., 2000; Schally et al., 2001; Massa et al., 2004; Di Bella 2005; Arena et al., 2007; Guillermet-Guibert et al., 2007; Di Bella 2008; Lee et al., 2008; Verhoef et al., 2008; Vieira Neto et al., 2008; Volante et al., 2008; Ben-Shlomo et al., 2009; Di Bella et al., 2009 ; Bellyei et al., 2010].
In molti tumori, non solo in quelli neuroendocrini, è stata documentata un’espressione recettoriale per la somatostatina [Moertel et al., 1994; Sestini et al., 1996; Kogner et al., 1997; Briganti et al., 1997; Van Eijck et al., 1998; Borgström et al., 1999; Friend et al., 2000; Albérini et al., 2000; Florio et al., 2000; Cattaneo et al., 2000; Steták et al., 2001; Orlando et al., 2001; Faggiano et al., 2008; Florio et al., 2008; Fusco et al., 2008; Kwekkeboom et al., 2008; Hubalewska-Dydejczyk et al., 2008; Ioannou et al., 2008; Khanna et al., 2008; Li et al., 2008; Corleto et al., 2009; Edelman et al., 2009; Hassaneen et al., 2009; He et al., 2009; Laklai et al., 2009; Luboldt et al., 2009; Pisarek et al., 2009; Ruscica et al., 2010].
È dimostrato anche il rapporto causale eproporzionale tra espressione recettoriale del GH (di cui la SST è l’antitodo biologico) e induzione e progressione tumorale [Friend et al., 2000; Zeitler et al., 2000; Gruszka et al., 2001], rilevando istochimicamente concentrazioni di GHR nettamente superiori nei tessuti tumorali rispetto a quelli sani. E’ pertanto noto, e ampiamente documentato, il potente ruolo mitogeno del GH, ed il fatto che l’indice proliferativo e la velocità di progressione delle popolazioni neoplastiche risulti direttamente proporzionale all’espressione recettoriale del GH stesso [Lincoln et al., 1998].
La proliferazione cellulare è strettamente dipendente dalla Prolattina [Bonneterre et al., 1990; Di Bella et al., 1997; Di Bella et al., 1998;Tada et al., 1999; Gruszka et al., 2001; Di Bella 2005; Florio et al., 2008; Mouton 2008; Di Bella 2009;] dal GH, massimo fattore di crescita [De Souza et al., l974; Di Bella et al., 1979; Di Bella et al., 1997; Di Bella L et al., 1998; Lincoln et al., 1998; Friend et al., 2000; Barnett et al., 2003; Anthony et al., 2009;] e da molecole mitogene GH dipendenti, da esso positivamente regolate, come EGF, FGF, HGF, IGF1-2, NGF, PDGF, TGF, VEGF [Szepesházi et al., 1999; Murray et al., 2004; Sall et al., 2004; Di Bella 2009; Hagemeister et al., 2008; Di Bella et al., 2009; Taslipinar et al., 2009] oltre che da fattori di crescita prodotti dall’apparato gastrointestinale, come VIP, CCK, G [Kath et al., 2000].
Sia la proliferazione cellulare fisiologica, che quella neoplastica, avvengono per mezzo di queste stesse molecole, che la cellula neoplastica utilizza, però, in rapporto esponenziale rispetto a quella sana. La perdita di differenziazione e la proliferazione incontrollata, anche se in misura diversa, caratterizzano tutte le neoplasie.
L’impiego della somatostatina e analoghi, agendo sulla crescita, denominatore comune a ogni tumore, deve trovare indicazione razionale in ogni neoplasia [Di Bella et al., 1979; Di Bella et al., 1981; Di Bella et al., 1997; Pollak et al., 1997; Di Bella et al., 1998; Pawlikowski et al., 1998; Friend et al., 2000; Lachowicz et al., 2000; Friend et al., 2000; Schally et al., 2001; Massa et al., 2004; Di Bella 2005; Arena et al., 2007; Guillermet-Guibert et al., 2007; Di Bella 2008; Lee et al., 2008; Verhoef et al., 2008; Vieira Neto et al., 2008; Volante et al., 2008; Ben-Shlomo et al., 2009; Di Bella et al., 2009 ; Bellyei et al., 2010].
In molti tumori, non solo in quelli neuroendocrini, è stata documentata un’espressione recettoriale per la somatostatina [Moertel et al., 1994; Sestini et al., 1996; Kogner et al., 1997; Briganti et al., 1997; Van Eijck et al., 1998; Borgström et al., 1999; Friend et al., 2000; Albérini et al., 2000; Florio et al., 2000; Cattaneo et al., 2000; Steták et al., 2001; Orlando et al., 2001; Faggiano et al., 2008; Florio et al., 2008; Fusco et al., 2008; Kwekkeboom et al., 2008; Hubalewska-Dydejczyk et al., 2008; Ioannou et al., 2008; Khanna et al., 2008; Li et al., 2008; Corleto et al., 2009; Edelman et al., 2009; Hassaneen et al., 2009; He et al., 2009; Laklai et al., 2009; Luboldt et al., 2009; Pisarek et al., 2009; Ruscica et al., 2010].
È dimostrato anche il rapporto causale eproporzionale tra espressione recettoriale del GH (di cui la SST è l’antitodo biologico) e induzione e progressione tumorale [Friend et al., 2000; Zeitler et al., 2000; Gruszka et al., 2001], rilevando istochimicamente concentrazioni di GHR nettamente superiori nei tessuti tumorali rispetto a quelli sani. E’ pertanto noto, e ampiamente documentato, il potente ruolo mitogeno del GH, ed il fatto che l’indice proliferativo e la velocità di progressione delle popolazioni neoplastiche risulti direttamente proporzionale all’espressione recettoriale del GH stesso [Lincoln et al., 1998].
E’ documentata l’inibizione di vari oncogeni, tra cui MIC da parte della SST e degli altri componenti del MDB [Degli Uberti et al., 1991; Peverali et al., 1996; Sun et al., 2002; Gumireddy et al., 2003; Durand et al., 2008; Aktas et al., 2009].
Tra i noti fattori causali dell’oncogenesi vi sono anche i danni cromosomici che comportano, in varia misura, inattivazioni di geni oncosoppressori: CD44, Bcl-2, P53, oltre che delle Caspasi 3-8, elementi chiave della cascata apoptotica. La regolazione negativa degli oncosopressori è antagonizzata da componenti del MDB come l’Ac. Retinoico, che inibisce l’inattivazione delle caspasi [Piedrafita et al., 1997; Takada et al., 2001; Jiang et al., 2008] e la MLT che preserva dalla degradazione P53 e Bcl-2 [Mediavilla et al., 1999].
L’inattivazione degli oncosoppressori può avvenire contemporaneamente all’amplificazione di oncogeni come il gene N-myc e il protooncogene TRK, considerati una delle cause citogenetiche neoplastiche.
Componenti del MDB come la STT e i retinoidi [Giannini et al., 1997; Witzigmann et al., 2008; Quan et al.,2008] antagonizzano la spinta proliferativa di queste molecole. Tra i fattori patogenetici, anche l’alterazione del sistema ligando–recettore GF-TRK, e l’alterata risposta allo stimolo differenziante, sono efficamente contrastati dai retinoidi [Hassan et al., 1990; Giannini et al., 1997; Peverali et al., 1996; Voigt et al., 2000, Kulikov et al., 2007; Beijersbergen et al., 2009; Wu et al., 2009; Witzigmann et al., 2008; Beijersbergen et al., 2009].
La differenziazione è potenziata sinergicamente da altri componenti del MDB come MLT [Cos et al., 1996; Garcia-Santos et al., 2006; McMillan et al., 1999], Vit D3 [Lange et al., 2007; Gocek et al. 2009], Vit E [Turley et al., 1995; Swettenham 2005], Vit C [Carosio et al., 2007], Condroitinsolfasto [Batra et al., 1997; Liang et al., 2009].
E’noto in che maniera l’asse GH-IGF1 abbia una determinante influenza sullo sviluppo biologico neoplastico [Murray et al., 2004].
Gli IGFR rispondono mitogenicamente a IGF.
L’effetto soppressivo della SST e analoghi sui livelli sierici di IGF1 è sia diretto, attraverso l’inibizione del gene di IGF [Cascinu et al., 1997] che indiretto, mediante
la soppressione del GH e pertanto della sua induzione epatica di IGF1 [Sall et al., 2004; Murray et al., 2004; Taslipinar et al.,2009].
Le cellule neoplastiche sono caratterizzate, anche se in misura diversa, da vari livelli di espressione dei recettori tirosinkinasici.
L’attività Proteinchinasica è efficacemente inibita dalla SST e analoghi [Reardon et al., 1996; Pawlikowski et al. 1998; Lachowicz-Ochedalska et al., 2000; Cattaneo et al., 2000; Florio et al.2001; Massa et al., 2004; Lee et al., 2008; Florio et al., 2008].
Anche l’espressione di TRK-B e l’amplificazione di N-Myc, insieme ad elevata attività telomerasica, comuni a diverse neoplasie, sono negativamente regolate dalla SST [Degli Uberti et al., 1991; Sun et al., 2002; Durand et al., 2008].
Antidoti biologici del GH, come Somatostatina e analoghi, riducono l’espressione e la trascrizione di fattori di crescita altamente mitogeni, come IGF 1-2 [Sall et al., 2004], FGF [Held-Feindt et al., 1999], VEGF [Albini et al., 1999; Vidal et al., 2000].
E’ documentata anche l’attività inibitoria della SST su un altro potente fattore di crescita mitogeno, l’EGF [Watt et al., 2009], attraverso molteplici meccanismi quali l’inibizione, dose dipendente, della fosforilazione tirosinica indotta dall’attivazione di EGFR da parte di EGF [Mishima et al.,1999], la riduzione di EGFR nelle cellule tumorali [Szepesházi et al., 1999] la riduzione dell’espressione di EGF [Held Feind et al.,2001], l’abbattimento della concentrazione plasmatica di EGF [Cascinu et al.,1997;Mishima et al., 1999; Szepesházi et al., 1999; Held-Feindt et al., 1999].
Somatostatina e analoghi estendono la loro regolazione negativa ai recettori dei già citati fattori di crescita, con evidenti riflessi antiangiogenici e antiproliferativi [Manni
et al., 1989; Barrie et al., 1993; Klijn et al., 1996; Pollak et al., 1997; Pawlikowski et al., 1998; Mishima et al., 1999; Lachowicz-Ochedalska et al., 2000; Friend et al.,
2000; Schally et al., 2001; Watson et al., 2001; Schally et al., 2003; Massa et al., 2004; Di Bella 2005; Arena et al., 2007; Guillermet-Guibert et al., 2007; Bocci et al.,
2007; Di Bella 2008; Lee. 2008; Di Bella et al.,2009].
E’ ormai assodato che la progressione neoplastica è strettamente dipendente dall’angiogenesi, e che quest’ultima ne rappresenta una fase obbligata ed essenziale.
L’acquisizione di un fenotipo angiogenico è decisivo per l’espansione del tumore[Longo 2002].
Somatostatina, e analoghi, regolano negativamente gli “induttori angiogenici”, e tutte le fasi dell’angiogenesi [Jia et al., 2003; Kunert-Radek et al., 2008] come la cascata dei monociti [Wiedermann et al., 1993], l’interleukina 8, la Prostaglandina E 2 e il VIP, l’Ossido-Nitrico-Sintasi endoteliale (e-Nos) [Florio et al., 2003] oltre ai fattori di crescita il cui sinergismo è essenziale per l’angiogenesi stessa, come il VEGF-A [Cascinu et al., 2001; Mentlein et al., 2001], TGF, [Murray et al,. 2004; Hagemeister et al., 2008], FGF, HGF [Jia et al,. 2003; Hagemeister et al., 2008], PDGF [Cattaneo et al., 1999]. L’inibizione dell’angiogenesi indotta dalla SST è sinergicamente e fattorialmente potenziata dagli altri componenti del MDB, quali MLT [Lissoni et al., 2001], Retinoidi [Majewski et al., 1994; McMillan et al., 1999; Kini et al., 2001; Liu et al., 2005], Vit D3 [Mantell et al., 2000; Kisker et al., 2003], Vit E [Shklar et al., 1996; Tang et al., 2001], Vit C [Ashino et al., 2003], inibitori prolattinici [Turner et al,. 2000], componenti della matrice extracellulare [Ozerdem et al., 2004; Liu et al., 2005].
Ugualmente documentato è l’effetto citostatico, antiproliferativo, antimetastatico della Somatostatina [Di Bella et al., 1979; Di Bella et al., 1981; Kogner et al., 1997; Di Bella et al., 1997; Schally et al., 1998; Di Bella et al., 1998; Orlando et al., 2001; Schally et al., 2003; Di Bella 2005; Arena et al., 2007; Krysiak et al., 2006; Guillermet-Guibert et al., 2007; Barbieri et al., 2008; Colucci et al., 2008; Di Bella 2008; Gambini et al., 2008; Li et al., 2008; Watt et al., 2008; Shima et al., 2008; Quan et al.,2008; Van Keimpema et al.,2008 ;Chen et al., 2009; Di Bella et al., 2009; Liu et al.,2009; Oberg et al., 2009; Hauser et al., 2009; Jia et al., 2009; Kaprin et al., 2009; Songgang et al., 2009; Nakashima et al., 2009; Zou et al., 2009; Ruscica et al., 2010].
L’effetto citostatico, antiproliferativo, antimetastatico della Somatostatina è efficacemente sinergizzato dagli altri componenti del MDB :
Retinoidi [Di Bella et al., 1979; Di Bella et al., 1981; Hassan et al., 1990; Di Bella et al., 1997; Onogi et al., 1998 ; Di Bella et al., 1998; Peverali et al., 1996; Piedrafita et al., 1997; Voigt et al., 2000; Di Bella G. 2005; Witzigmann et al., 2008; Di Bella 2008; Schilling et al., 2008; Pyronnet et al., 2008; Di Bella et al., 2009; Nakagawa et al., 2009]
MLT [Di Bella et al., 1979; Di Bella et al., 1981; Kvetnoĭ et al., 1986; Maestroni et al., 1996; Cos et al., 1996; Di Bella et al., 1997; Di Bella et al., 1998; Bartsch et al., 1999; Mediavilla et al., 1999; Cos et al., 2000 ; Di Bella 2005 ; García-Santos et al., 2006; Mc Millan et al.,2007; Di Bella 2008; Srinivasan et al., 2008; Bonofiglio etal., 2009; Di Bella et al.,2009; Srirajaskanthan et al., 2009],
Vitamina D3 [Barroga et al., 2000; Campbell et al., 2000; Jensen et al., 2001 ; Stio et al., 2001; Di Bella G. 2005; Di Bella G. 2008; Gocec et al., 2009; Di Bella G. et al.,2009; ],
Cabergolina e Bromocriptina (inibitori prolattinici) [Di Bella et al.,1979; Di Bella et al.,1981; Klijn et al., 1989; Klijn et al., 1996; Di Bella et al., 1997; Di Bella et al., 1998; Lissoni et al.,2000; Gruszka et al., 2001; Frontini et al.,2004; Di Bella 2005; Senogles et al., 2007; Di Bella 2008; Mouton et al., 2008; Di Bella et al.,2009; Srirajaskanthan et al., 2009],
Galattosamina solfato, Calcio [Batra et al., 1997; Di Bella 1997; Di Bella et al., 998; Pumphrey et al,. 2002; Di Bella 2005, Di Bella 2008; Di Bella et al., 2009],
Vit E [Cameron et al., 1979; Di Bella et al., 1979; Turley et al., 1995; Shklar et al., 1996; Di Bella et al., 1997; Di Bella et al., 1998; Israel et al., 2000; Malafa et al., 2002; Neuzil et al., 2002 ; Di Bella 2005; Di Bella 2008; Di Bella et al., 2009],
Vit C [Murata et al., 1982; Head et al., 1998; Steták et al., 2001; Di Bella 2005; Carosio et al., 2007; Florio et al., 2008, Di Bella 2008; Di Bella et al., 2009] .
La letteratura ha pertanto confermato i sinergici meccanismi d’azione antineoplastici differenzianti, citostatici, antiproliferativi, antiangiogenici e antimetastatici di tutti i componenti del MDB.
Senza l’ apporto dell’ormone della crescita [GH] e dei Fattori di Crescita [GF] prodotti dai tessuti per azione del GH, e quindi strettamente GH-dipendenti, non esiste crescita fisiologica o tumorale.
Le mutazioni cellulari avvengono per varie cause, di ordine fisico, chimico, infettivo. Diversi componenti del MDB (MLT, Vit D3, C, E, Retinoidi, componenti della ECM) hanno un effetto differenziante.
Nella crescita dei tumori ormono-dipendenti, intervengono anche l’estrogeno (nei tumori della mammella e utero), e il testosterone (nel carcinoma prostatico e dei testicoli).
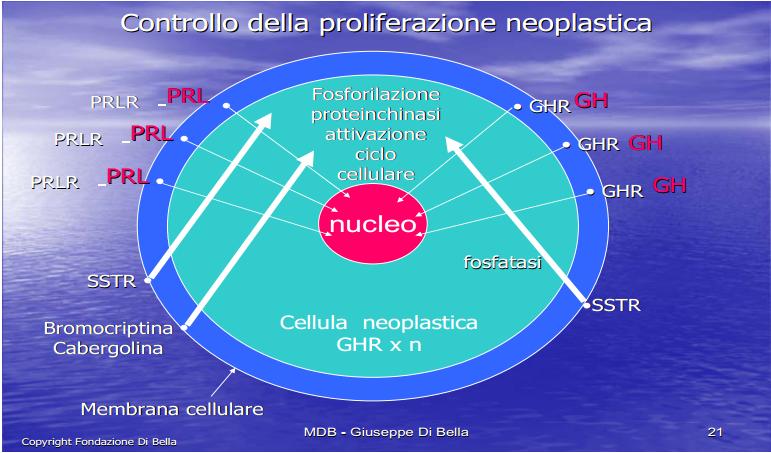
Meccanismo recettoriale della crescita
Le molecole GH, GF e PRL attivando i rispettivi recettori di membrana GHR, GFR e PRLR, avviano reazioni chimiche di fosforilazione trasferendo il segnale dalla membrana cellulare al nucleo. Più alta è la quantità di GHR in una cellula tumorale, maggiore è la sua capacità di utilizzare il GH, e pertanto di crescere, sia localmente, che di espandersi anche a distanza.
E’ ampiamente dimostrato il rapporto dose-dipendente tra espressione recettoriale del GH nelle cellule tumorali, e la loro capacità e velocità di proliferazione ed espansione sia locale che a distanza, migrando e producendo metastasi.
Per questo, essendo definitivamente e scientificamente documentato che il tumore è crescita, e che questa crescita dipende da GH, GF e PRL, l’ovvio obiettivo terapeutico primario della cura di qualsiasi tumore, non può conseguentemente prescindere dall’inibizione di GH, GF e PRL, mediante Somatostatina e gli inibitori prolattinici Cabergolina e/o Bromocriptina.
Pertanto l’inibizione della crescita tumorale attraverso il blocco dell’ormone della crescita, per mezzo del suo antidoto biologico, la Somatostatina [SST], segue una logica semplice, lineare, comprensibile e matematica.
| L’ormone della crescita GH, a diretto contatto col rispettivo recettore GHR, a livello della membrana cellulare (in blu). Il contatto avvia una reazione di trasduzione e amplificazione di segnale al nucleo (in rosso). Le reazioni sono di fosforilazioni protein-tirosinchinasiche. Queste reazioni sono bloccate dalla somatostatina (SST) che, attivando il recettore SSTR, avvia sistemi enzimatici OPPOSTI di fosfatasi, che inattivano la catena di fosforilazioni, protein-tirosin chinasiche, inibendo la proliferazione neoplastica. Questa azione antitumorale diretta della SST sulla cellula tumorale, si somma a quella indiretta, altrettanto potente, consistente nell’abbattimento della concentrazione ematica del GH e conseguentemente di GF. |
Lo stesso concetto, lo stesso razionale terapeutico, viene applicato al blocco farmacologico della Prolattina mediante i relativi inibitori, quali la Bromocriptina e la Cabergolina. Lo stesso concetto, lo stesso razionale terapeutico, viene applicato in oncologia al blocco degli estrogeni ed androgeni nei rispettivi tumori ormono dipendenti . Ma l’oncologia non ha ancora chiara la necessità di estendere lo stesso concetto all’inibizione dei due più potenti oncogeni ubiquitari , GH , GF ( Gh dipendenti) e PRL.
L’oncologia continua a trastullarsi col recettore della somatostatina (SSTR) vincolando e limitando il suo impiego alle situazioni in cui viene individuato il suo recettore nelle cellule tumorali. L’esame più frequente per questa ricerca è l’Octreoscan. Per questa indagine viene iniettata in vena somatostatina di sintesi, spesso Octreotide, radiomarcata, e mediante scintigrafia viene studiata la presenza, nei tessuti, di SSTR. Questa tecnica è poco affidabile in quanto non sempre è in grado di evidenziare neppure 2 dei sette recettori della somatostatina, il 2 e il 5, ed ha dimostrato di avere un’alta percentuale di falsi
negativi. Infatti in molte situazioni di Octreoscan completamente negativo, mediante indagini più affidabili, come l’immunoistochimica e la transcriptasi inversa, è stata accertata la presenza di SSTR [Schaer et al., 1997; Van Eijck et al., 1998; Held-Feindt et al., 1999; Mishima et al., 1999; Pinzani et al., 2001; Watson et al., 2001;Barnett et al., 2003].
La convinzione che l'Octreoscan serva per saggiare l'utilità della somatostatina è pertanto superata. La negatività dell’Octreoscan non condiziona minimamente il razionale dell’indicazione antitumorale della somatostatina per molteplici motivi: tutte le cellule tumorali hanno indici di crescita dose-dipendenti rispetto all’espressione del recettore dell’ormone della crescita [Lincoln et al., 1998].
Il GH inoltre promuove la crescita tumorale anche con un meccanismo indiretto, l’induzione dei “Fattori di crescita” (GF), molecole fortemente mitogene che i
tessuti possono produrre, se attivati dal GH.
In assenza dell’ormone della crescita (GH) nessun tessuto può produrre i Fattori di crescita.
Pertanto il GH ha un essenziale, forte e duplice ruolo mitogeno:
- Diretto sulla crescita della cellula tumorale, mediante attivazione dei rispettivi recettori di membrana GHR,
- Indiretto attraverso l’induzione nei tessuti di fattori di crescita (GF), responsabili di una formidabile accelerazione della crescita neoplastica.
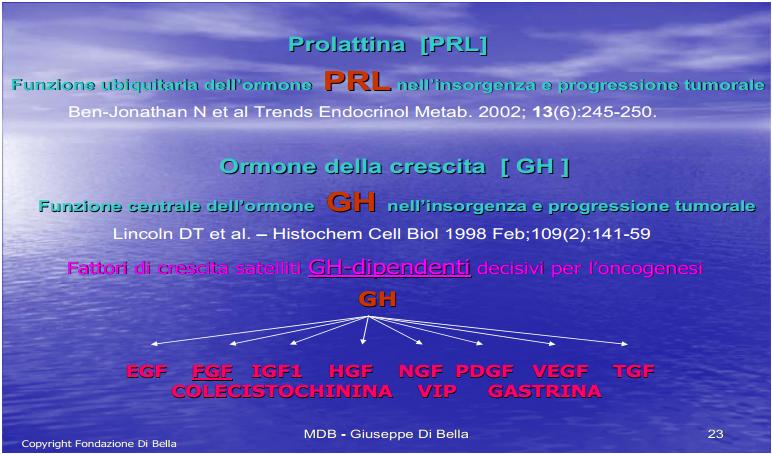
| Tra i fattori di crescita GH dipendenti che svolgono un ruolo primario nell’induzione e progressione neoplastica: EGF Fattore di crescita epidermico , FGF fibroblastico, HGH di derivazione dagli epatociti, IGF 1-2 prodotto dal fegato, NGF di derivazione dalle cellule nervose, PDGF, prodotto dalle piastrine, VEGF del tessuto vascolare, TGF fattore di trasformazione ecc… |
Regolando negativamente con la somatostatina il GH, e i GF dipendenti, si agisce pertanto direttamente contro la crescita tumorale in presenza, o meno, di recettori per la somatostatina (SSTR) a livello delle cellule neoplastiche. La presenza di SSTR sulla membrana delle cellule neoplastiche può accelerare o intensificare la risposta alla SST , ma questa risposta comunque avverrebbe per le suddette ragioni.
La prassi di vincolare l’utilizzo terapeutico della somatostatina unicamente al riscontro del suo recettore nella cellula neoplastica risulta pertanto irrazionale. Inoltre, anche quando non vengono evidenziati recettori della somatostatina (SSTR) nel tumore, essi sono comunque e sempre reperibili nei vasi peritumorali.
La SST pertanto, bloccando GH e relativi GF, costituisce la più potente inibizione della proliferazione tumorale, e rappresenta una condizione necessaria ed essenziale, anche se non sufficiente, nella cura di tutti i tumori, con o senza presenza di recettori della somatostatina, limitando, quindi, anche le indicazioni terapeutiche degli anticorpi monoclonali.
Con gli anticorpi monoclonali si inibiscono le chinasi attivate dai GF, ma è ampiamente documentato che la SST inibisce l’espressione genica di
tutti i GF, ne blocca la trascrizione, ed estende il blocco all’espressione e trascrizione dei rispettivi recettori. Già abbattendo, con la SST, il tasso plasmatici di GH, si sottrae la molecola base necessaria per la sintesi dei GF.
La tossicità degli anticorpi monoclonali è dovuta al fatto che il recettore della membrana cellulare su cui agisce l’anticorpo monoclonale non è costituito dal solo gene del GF da bloccare, ma da un’intera famiglia di geni, tutti inattivati dall’anticorpo monoclonale. E’ proprio il blocco indesiderato, ma inevitabile, di tutti gli altri geni collegati al recettore del GF, che induce tossicità. Spesso l’anticorpo monoclonale viene impiegato contemporaneamente o sequenzialmente alla chemio, con una logica molto difficilmente comprensibile: la CA è infatti citotossica e citolitica, lisa e/o intossica le cellule e, nella grande percentuale di cellule che non elimina, sovverte più o meno gravemente lo strato superficiale, più fragile ed esposto della cellula, la membrana
cellulare, sede dei recettori su cui agiscono gli anticorpi monoclonali, eliminando così, o sovvertendo, quei siti recettoriali su cui dovrebbe agire l’AM stesso, oppure denaturando gravemente i potenziali di membrana, i canali ionici e la catena di trasduzione del segnale al nucleo . E’ come se si mettesse fuori uso l’interruttore, o si tagliasse il filo che lo collega alla lampadina, e poi si pretendesse di accendere la luce .
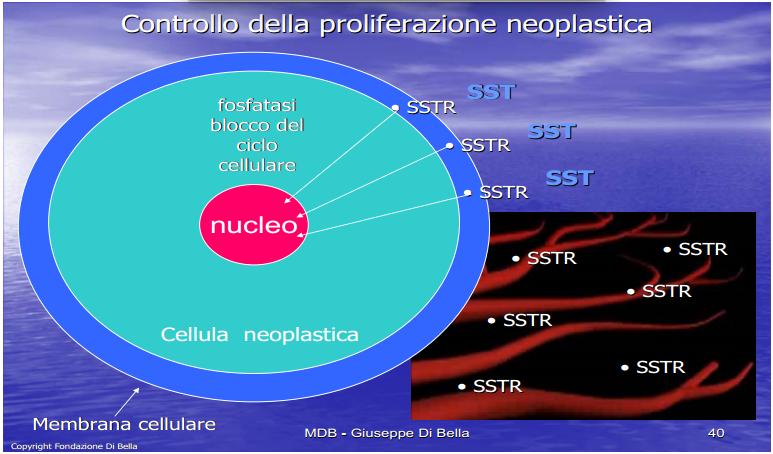
| In blu la membrana cellulare con i recettori della somatostatina SSTR , in verde il citoplasma della cellula all’interno della membrana, in esso avvengono le reazioni chimiche attivate dal contatto tra la SST (ligando), e il recettore SSTR. Queste reazioni (fosfatasi), segnate con la freccia bianca, bloccano le reazioni di fosforilazione protein-tirosin chinasica della proliferazione tumorale indotte da GH e GF. Sul lato DX della figura la rappresentazione schematica dei vasi sanguigni che circondano il tumore, dandogli apporto nutritivo, in cui è stata documentata un’elevata e costante espressione di SSTR. |
I vasi sanguigni peritumorali presentano infatti, costantemente, una concentrazione di recettori (SSTR) che, se attivati dalla somatostatina, regolano negativamente l’angiogenesi e, conseguentemente, la progressione neoplastica. E’ pertanto documentato che, anche nei casi in cui nella cellula neoplastica non viene riscontrato alcun SSTR, la somatostatina agisce direttamente ed efficacemente bloccando la crescita tumorale attraverso l’inibizione dell’angiogenesi e, senza la quale non può svilupparsi alcun tumore. Ad esempio nelle cellule del sarcoma di Kaposi, in cui è stata evidenziata l’assenza completa di SSTR, la crescita è completamente bloccata dalla somatostatina. Nel sarcoma di Kaposi, infatti, è stata riscontrata un’alta densità
di SSTR nei vasi sanguigni peritumorali, per cui l’effetto citostatico è conseguente a quello antiangiogenico da parte della SST. ( Albini , et al., 1999)
Fino al momento in cui le cellule che costituiscono il primo aggregato tumorale di pochi millimetri non riescono a crearsi un proprio sistema di vasi sanguigni
(Angiogenesi neoplastica), esse crescono con estrema lentezza e sono destinate a non superare le dimensioni di qualche millimetro, rimanendo allo stadio di “cancro in situ”. L’espansione tumorale avviene solo quando il tumore realizza l’angiogenesi, riesce cioè a costruirsi una rete di vasi sanguigni per assicurarsi l’apporto di sostanze nutritive e l’eliminazione di scorie metaboliche.
La letteratura ha documentato che tutti i passaggi dell’angiogenesi sono negativamente regolati dalla somatostatina e dai suoi analoghi e, anche se in misura minore, da tutti gli altri componenti del MDB.
Se l’espansione neoplastica ha nell’angiogenesi un passaggio obbligato, e se l’angiogenesi è totalmente inibita dalla somatostatina, è ulteriormente chiarita e documentata la sua indicazione in tutti i tumori, in presenza o meno, di SSTR. Anche le situazioni locali di anossia e acidosi favoriscono l’angiogenesi , e in buona parte sono corrette dal miglioramento degli scambi emotissutali indotto dai component differenzianti del MDB.
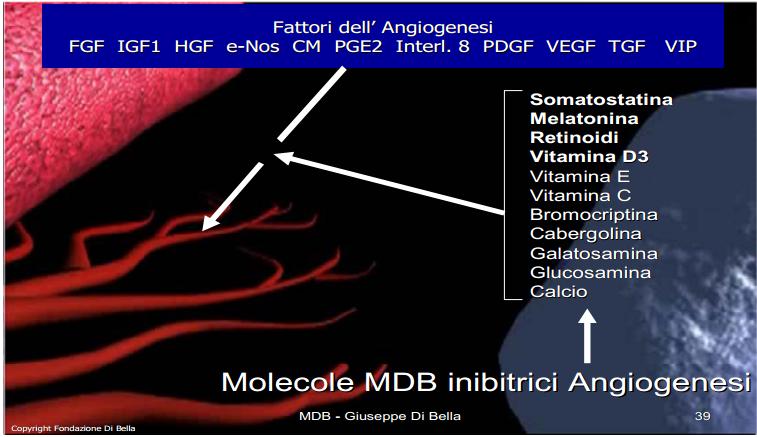
Molecole che concorrono a promuovere l’angiogenesi inibite sinergicamente dalla somatostatina e da ogni singolo componente del MDB: Ossido-Nitrico-Sintasi endoteliale (NOSe) Interleuchina 8 ( Il8) Chemiotassi dei Monociti GH indotta (C. M) Prostaglandina 2 (PG2) Fattore fibroblastico di crescita(FGF) Fattore di crescita di derivazione dagli epatociti( HGF) Fattore di crescita di derivazione epatica (IGF 1-2) Fattore di crescita di derivazione piastrinica (PDGF) Fattore di crescita vascolare (VEGF) Fattore di crescita di trasformazione (TGF) |
CONTRASTO ALLA TENDENZA MUTAGENA DEL FENOTIPO NEOPLASTICO
Inibizione delle mutazioni della cellula tumorale
L’altro aspetto fondamentale della progressione neoplastica, e pertanto obiettivo della razionalità terapeutica del MDB, è costituito dalle mutazioni delle cellule tumorali, perché ad ogni mutazione la cellula seleziona e trattiene una serie di vantaggi. Le proprietà differenzianti di componenti del MDB come Melatonina, Retinoidi, VIT E, C, D3, e componenti della matrice extracellulare (ECM), si oppongono alla spiccata tendenza mutagena del fenotipo neoplastico
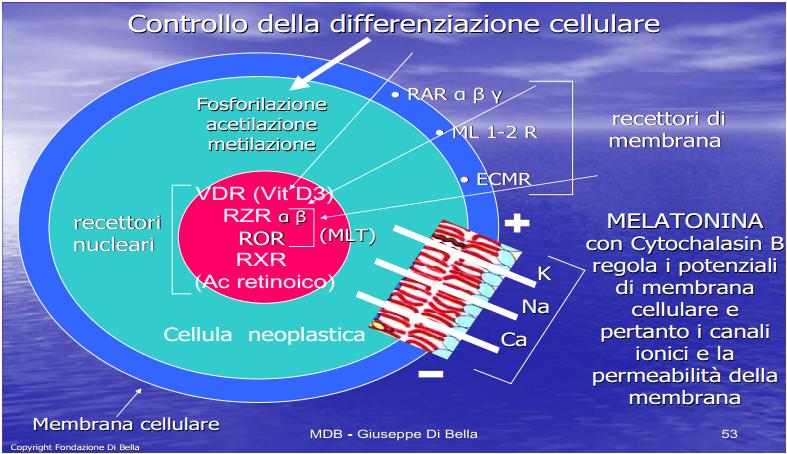
| Sulla membrana cellulare, in azzurro, sono riportati i siti recettoriali differenzianti: RAR dei retinoidi, con 3 sottogruppi (alfa beta e gamma), MELR della melatonina, ECMR della matrice extracellulare. Nel nucleo, in rosso, sono riportati i recettori nucleari RXR dell’Acido Retinoico, VDR della vitamina D3, ROR e RZR alfa e beta della Melatonina. I ligandi di questi recettori, sia di membrana che nucleari, sono componenti del MDB, e associano ad una risposta sinergica differenziante il potenziamento antiproliferativo della SST e inibitori prolattinici. Se attivati tempestivamente, e sinergicamente, tutti questi blocchi recettoriali delle mutazioni e della proliferazione neoplastica, difficilmente possono essere superati. Nella zona di membrana ingrandita con i segni + e - alle estremità, sono localizzati i canali ionici del calcio, sodio e potassio, di vitale importanza per l’equilibrio biologico e il contrasto al tumore: modulati dalla melatonina, attraverso il controllo dei potenziali di membrana, realizzano l’effetto differenziante mediante l’attivazione di reazioni di Fosforilazione, Metilazione , Acetilazione . |
Gli obiettivi strategici di una cura antiblastica, pertanto, non possono prescindere dal controllo delle mutazioni, che rappresentano una caratteristica essenziale e un denominatore comune delle cellule tumorali, non meno della citata dipendenza per la crescita da GH, PRL, e GF.
La cellula tumorale è caratterizzata da una frequenza di mutazioni crescente e segue, nella sua progressione, un programma predefinito di sopravvivenza ereditato dai batteri [Radman et al., 1975] (cui è stato trasferito dai procarioti) definito da Radman “SOS”, che è represso, ma presente, nella cellula sana, ed al quale essa accede in condizione di stress acuto.
Questo programma di sopravvivenza, dà avvio a un percorso predefinito che consente alla cellula, divenuta neoplastica, di adattarsi con grande rapidità ed efficacia alle condizioni avverse con una progressione modulata da un meccanismo evolutivo predeterminato.
Il paradigma ancora dominante, i canoni ufficiali dell’oncologia, non hanno ancora recepito questo essenziale aspetto dell’evoluzione neoplastica, ormai necessario per una comprensione della biologia oncologica e per dare una lettura in termini evoluzionistici della progressione della malattia tumorale (da non confondere con una concezione darwinista). I protagonisti dell’evoluzione in realtà sono la selezione naturale e la variazione genetica. La selezione naturale agisce sulla variazione genetica conferendo un vantaggio evolutivo a fenotipi e genotipi che meglio si sono adattati all’ambiente.
La fonte della diversità genetica è la mutazione nelle sequenze del DNA, e la mutazione è un fenomeno, per definizione, totalmente casuale, integralmente gestito dal caso.
Quindi nell’ambito dell’evoluzione, in cui agiscono le mutazioni e la selezione naturale, è chiaro che tutto viene pilotato dal caso.
Naturalmente anche il cancro segue questa prassi evolutiva, e sicuramente è un processo di evoluzione somatica totalmente pilotato dal caso quello che porta alla carcinogenesi. Nell’uomo essa è un processo genetico, la cui dinamica è regolata dall’interazione fra mutazione, selezione, e i meccanismi di omeostasi antiblastica dell’organizzazione tissutale, propria degli organismi complessi pluricellulari superiori e ovviamente ad essi limitata. L’evoluzione di una cellula verso la malignità ha inizio con una o più mutazioni casuali. Queste mutazioni conferiscono ovviamente alla cellula un vantaggio in termini proliferativi e dunque vengono in qualche modo trattenuti dalla selezione. Quindi la lettura attuale della malattia tumorale è in termini evolutivi. Naturalmente l’accumulazione di mutazioni produrrà ondate successive di espansioni clonali.
Secondo il paradigma prevalente della visione ortodossa del cancro, esso è una malattia genetica, originata soprattutto dalla mutazione di 2 classi di geni, gli oncogeni e gli oncosoppressori, pertanto da mutazioni dei geni che regolano la differenziazione e la crescita, direttamente coinvolti nell’evoluzione, e di quelli preposti a mantenere l’integrità del DNA, deputati alla sorveglianza della fedeltà della sintesi del DNA, e alla sua riparazione mediante i molteplici meccanismi apparsi nel corso dell’evoluzione.
Tra i geni che regolano l’omeostasi antiblastica un ruolo fondamentale è svolto da quelli che generano l’apoptosi. Ogni qualvolta si presenti una mutazione in questi geni, una o più mutazioni, si assiste ad una progressione della malattia tumorale. Così quando si verifica una mutazione soprattutto nei geni di riparazione del danno del DNA, si verifica quella che è stata definita instabilità genetica, cioè il fenotipo mutante. Una semplice mutazione di una cellula sana, non riuscirebbe a spiegare questo accumulo di mutazioni e quindi si invoca la presenza di un fenotipo molto più instabile.
Probabilmente c’è un errore di posizione sul concetto di instabilità genetica. Nella concezione di Radman (basata sul sistema di sopravvivenza definito “SOS” e sostenuta da Israel et al.,) i due attori fondamentali sono il gene LexA e il gene RecA e le relative proteine. Il gene LexA è un repressore trascrizionale, mentre il gene RecA è invece un regolatore positivo. Rimando alle pubblicazioni citate per approfondimenti.
In condizioni di stabilità il programma di sopravvivenza “SOS” non è attivo, esso è represso dal gene LexA. Il sistema “SOS” comprende circa una ventina di geni e quindi quando il DNA viene danneggiato o comunque la sopravvivenza della cellula è in pericolo, la proteina LexA in qualche modo viene inattivata dalla produzione di un’altra proteina, la RecA, ed è a questo punto che si attivano i geni. Sicuramente questo programma è stato messo a punto da mutazioni casuali, selezionate favorevolmente e trattenute dalla cellula che ha accesso a questa informazione in condizioni particolari.
Vi sono forti indizi per ritenere, con gli A.A. citati, che questo programma che è stato trattenuto dall’evoluzione, ed è presente negli eucarioti, sia stato trasmesso alle nostre cellule.
La ricerca di un programma “SOS” nelle cellule eucariote e negli organismi multicellulari come il nostro, ha già dato risultati positivi. Gli studi del professor
Israel, portano a ricercare omologie, tra le proteine e i geni del sistema “SOS” batterico e quelli trattenuti nelle nostre cellule [ Israel 1996 ]
Uno di questi geni è stato già identificato. C’è un’omologia molto marcata tra la proteina batterica RecA e una proteina presente nelle nostre cellule, la Rad .
Dunque abbiamo fondate ragioni di ritenere che il sistema “SOS”, anche in una sua versione molto più evoluta, possa esistere anche nelle nostre cellule. Ad un approfondito esame l’attuale paradigma dominante della visione della progressione maligna come totalmente gestita dal caso, cioè interamente prodotta da una somma di mutazioni successive, ma sempre casuali, non regge, per il carattere piuttosto prevedibile della progressione maligna. Ad eccezione degli eventi iniziali, sicuramente gestiti da casuali mutazioni, la progressione della malattia tumorale è sicuramente molto stereotipata, è la recita di un copione.
Le cellule tumorali acquisiscono con gradualità e progressione, crescenti proprietà e caratteristiche, ed “imparano” a svolgere tutta una serie di attività. Un fenotipo così caratterizzato, necessita di circa un migliaio di generazioni. Considerando che un tempo di generazione è di circa 48 ore, in un periodo così relativamente breve le cellule tumorali sono in grado di produrre una serie di fattori di crescita che le loro omologhe, non endocrine, non sanno sintetizzare. Le cellule tumorali esprimono dei recettori a questi fattori, che influenzano la proliferazione selettiva, limitata alle stesse popolazioni neoplastiche.
Esse inoltre acquisiscono sempre maggiori motilità e formabilità per meglio raggiungere i capillari e aumentare il proprio potenziale di metastasi, sanno inoltre acquistare capacità di sopravvivenza e di proliferazione in parenchimi anche diversi, e ricoprirsi di molecole che le mascherano al sistema immunitario. Successivamente sono in grado di secernere delle proteasi che, lisando le membrane, permettono una invasione per contiguità, oltre a indurre angiogenesi e immunodepressione locale e sistemica. In un lavoro pubblicato nel 2003 su “Nature” si documenta come una cellula di melanoma attaccata da un linfocita, sia in grado di produrre “apoptosi” nel linfocita; quindi le popolazioni neoplastiche raggiungono progressivamente la capacità di eliminare le cellule del sistema immunitario che tentano l’aggressione.
Per ultimo la cellula tumorale è in grado di modificare l’ambiente cellulare circostante, inducendo le cellule vicine a sostenere la propria proliferazione.
Il fatto stesso che siano agevolmente in grado di codificare i passaggi essenziali della progressione verso la malignità e di acquisire un graduale incremento di aggressività, proliferazione, adattamento, contraddice una visione evolutiva strettamente casuale della malattia tumorale.
Ci sono ulteriori aspetti che danno conforto a questa posizione, le sindromi paraneoplastiche, una sorta di cartina al tornasole della progressione verso la
malignità. Un dato significativo è costituito dal fatto che, se queste mutazioni fossero gestite dal caso, o meglio se la progressione fosse totalmente gestita dal caso, dovremmo assistere sia a mutazioni favorevoli, che sfavorevoli, o comunque neutre, rispetto all’evoluzione tumorale. In realtà questo non succede. Le sindromi paraneoplastiche documentano come la produzione di sostanze anomale, da parte della cellula tumorale, mostri sempre un’utilità biologica per il tumore, che produce soltanto sostanze che gli tornano utili [Israel, 1996].
Ciò è fortemente in contraddizione con l’idea oncologica ufficiale di una progressione casuale, perché in questo caso dovremmo assistere anche a produzione di sostanze (se è il caso che gioca) neutre, o comunque anche sfavorevoli, rispetto alla progressione tumorale. Esistono alcuni eventi genetici, caratterizzanti la progressione tumorale, che non corrispondono a delle mutazioni, ma sono semplici riattivazioni e repressioni o amplificazioni di geni, non mutati, ma silenti.
Questo inevitabilmente ci porta a concludere che sicuramente gli organismi multicellulari più evoluti, come il nostro, hanno ereditato parti di genoma dai batteri, come emerge chiaramente nei recenti lavori di genetica molecolare in cui si documenta che certi geni batterici si sono assolutamente conservati nelle nostre cellule.
Nell’evoluzione degli organismi pluricellulari verso una sempre maggiore complessità, il destino di ogni cellula si lega a quello della collettività a cui appartiene.
L’evoluzione verso la complessità, verso un organismo pluricellulare prevede una sorta di cooperazione della collettività cellulare e dunque l’introduzione di nuove regole; in questo senso l’evoluzione ha messo a punto una sorta di controprogramma o comunque di sistema, che controlla l’omeostasi tissutale, cosa che ovviamente non è possibile e necessaria in un ambiente batterico o unicellulare.
Questo è il sistema degli oncosopressori, che assicura l’omeostasi cellulare antiblastica, impedendo ad ogni singola cellula di affrancarsi e acquistare una propria autonomia, che potrebbe mettere a rischio l’intera collettività tissutale.
L’evoluzione ha prodotto questo sistema, sicuramente più giovane e quindi più imperfetto e con delle lacune, che è il sistema degli oncosoppressori.
Conseguentemente la ricerca non ha evidenziato negli eucarioti gli omologhi degli oncosoppressori, quindi abbiamo ragione di ritenere che gli ncosoppressori siano dei geni emersi evolutivamente più tardi. Tra gli oncosoppressori è di particolare interesse il gene p53, guardiano del genoma, direttamente coinvolto nell’attivazione di un programma cellulare fondamentale per l’omeostasi antiblastica, quello dell’apoptosi.
Mi sono dilungato sul programma di sopravvivenza di Radman per evidenziare, anche alla luce di queste acquisizioni, la razionalità dei criteri, dei meccanismi molecolari, degli obiettivi del MDB. Le ricerche di Radman, recepite e sviluppate dal Professor Israel, ed esposte dal Prof. Truc al I° Congresso Nazionale MDB del maggio 2004 [Di Bella 2005], e qui riportate, hanno dato maggiore consapevolezza del fatto che la proteiforme capacità di adattamento della cellula tumorale, la sua formidabile vitalità, capacità mutagena e di recupero, sconosciute alla biologia umana fisiologica, sono state gravemente sottovalutate. L’esatta e realistica valutazione dei pressoché illimitati potenziali biologici neoplastici porta ad una logica terapeutica esattamente conforme ai postulati e al razionale del MDB: solo un precoce attacco multiterapico sinergico e concentrico, senza discontinuità spazio-temporale può tenere testa,
contenere e prevalere su una forma di vita diversa e drammaticamente superiore alla fisiologica, con altissime capacità di adattamento, e di superamento, ad ogni singola condizione avversa la medicina possa opporle. La cellula neoplastica supera facilmente qualsiasi singolo ostacolo, per quanto efficace, pertanto solo la contemporanea attivazione di tutta una serie di blocchi alle mutazioni neoplastiche può impedire il più micidiale meccanismo di difesa della cellula tumorale, la mutazione. Solo l’effetto fattoriale sinergico dei componenti multiterapici differenzianti, citostatici e antiproliferativi del MDB, può contrastare ad un tempo la proliferazione esponenziale del fenotipo neoplastico e la sua elevatissima capacità mutagena, efficientissimo sistema difensivo difficilmente penetrabile. È necessario agire su elementi critici del processo neoplastico come la differenziazione, attraverso la contemporanea attivazione di molteplici bersagli recettoriali differenzianti come i 45VDR (recettori nucleari della Vit D3), RXR (recettore nucleare dell’ac.retinoico), RAR–alfa, beta,gamma (recettori di membrana dei retinoidi), Mel-1,2 RZR/ROR (recettori di membrana e nucleari della Melatonina). Al tempo stesso occorre sottrarre alla cellula neoplastica la maggiore varietà e la massima entità, concentrazione possibile di energia, rappresentata da GH, GF, PRL e nei tumori ormonodipendenti da estrogeni o androgeni. L’obiettivo si realizza sia inibendo l’increzione di GH ipofisario e relativo GF, con SST e analoghi, che quella di PRL con Bromocriptina e/o Cabergolina. Un’ampia rassegna della relativa letteratura (circa 2000 voci) è riportata nel volume “Il Metodo Di Bella“ [ Di Bella 2005]. La MLT esercita un particolare, determinante e multifunzionale effetto di regolazione negativa dell’angiogenesi, sia inibendone un essenziale componente, il PDGF, che
regolando (con meccanismo omeostatico di modulazione serotoninergica consentito anche dal suo legame di idrogeno con l’adenosina), il tasso trombocitemico, l’aggregazione piastrinica (sinergicamente all’Alfa MSH), il tono vasale e la permeabilità endoteliale (attraverso la modulazione di EDRF ed EDCF), fattori essenziali per la liberazione del PDGF.
Bersagli terapeutici innovativi del MDB sono anche l’omeostasi dell’ambiente in cui vive la cellula tumorale, la regolazione fisiologica dei potenziali di membrana cellulare, le membrane basali, dotate di documentata attività differenziante, le proteine di adesione, le fasce di contenimento dell’espansione neoplastica, tutta la matrice extracellulare, il trofismo ed efficienza di parenchimi e tessuti, e degli endoteli, con relativa riconduzione a livello fisiologico della permeabilità vasale, degli scambi e della perfusione emo-tissutale. Anche l’esaltazione dell’immunità mediante le formulazioni , posologia e dosaggi di MLT e dei componenti vitaminici impiegati è conseguita dal MDB.
Tra le finalità del MDB, il recupero a livello fisiologico dei ritmi biologici circadiani, sovvertiti nelle neoplasie, mediante la modulazione della biodisponibiltà degli indoli pinealici in un contesto di continuità terapeutica temporale, intesa come assedio continuativo di una cellula tumorale già sensibilizzata dai numerosi agenti differenzianti, alla quale vengono al tempo stesso sottratti ormoni e fattori di crescita, senza concederle (diversamente dai cicli chemioterapici) pause di recupero, il tutto integrato da minimali dosaggi apoptotici, non citotossici e non mutageni di chemioterapici, la cui tollerabilità è esaltata dalla MLT e dalle vitamine del MDB.
CASISTICA
Origine di questo studio retrospettivo osservazionale:
Origine di questo studio retrospettivo osservazionale:
In Italia dalla fine degli anni settanta un numero crescente di ammalati di tumore aveva scelto di curarsi col Metodo Di Bella (MDB), messo a punto e
progressivamente affinato e integrato dal medico e scienziato Prof. Luigi Di Bella, laureato anche in chimica e farmacia e docente universitario di fisiologia generale, fisiologia umana e biochimica, preferendolo alla chemioterapia. La tollerabilità della terapia e i positivi risultati in termini di sopravvivenza e di qualità di vita, portarono ad una sempre maggiore diffusione del MDB ( praticato da migliaia di pazienti), creando un grave contenzioso tra opinione pubblica e istituzioni sanitarie, che cercarono di eludere la sempre più pressante richiesta di erogazione gratuita del MDB con una sperimentazione di fase II, la cui progettazione, conduzione e conclusione, furono causa di aspre critiche e sconcerto, non solo nell’opinione pubblica, ma anche in ampi settori politici, medici e dell’informazione, fino a portare ad indagini della magistratura. Le anomalie di tale sperimentazione furono anche oggetto di oltre cinquanta interrogazioni parlamentari, pubblicate sulla Gazzetta Ufficiale del Parlamento Italiano. Indipendentemente dalle numerose e gravi anomalie documentate, e rilevate anche dalla letteratura internazionale .L’Istituto Superiore di Sanità italiano , responsabile della sperimentazione , ha sostenuto che: «E’ stato necessario effettuare uno studio senza gruppo di controllo perché, nella situazione esistente dall’inizio del 1998, ciò non era concepibile…».
Non era concepibile fare il tipo di studio che viene fatto in tutto il mondo, ed anche in Italia, correntemente (ma non per il MDB!?).
Questa argomentazione non tiene: infatti, quando la sperimentazione del MDB. è stata pubblicata sul British Medical Journal, l’editorialista Marcus Muller (fatto del tutto inusuale) ha criticato aspramente la progettazione dello studio, sostenendo che : «Gli autori (della sperimentazione) affermano anche che non avrebbero potuto condurre uno studio clinico randomizzato per ragioni etiche, ma queste ragioni non sono chiare. In realtà, c’è chi può sostenere che proprio il livello inferiore di progettazione dello studio è anti-etico».
Ed a proposito della mancanza del gruppo di controllo, ecco cosa afferma in una lettera al B.M.J. il ricercatore Rey M.D.: «Con cosa è stata comparata la terapia Di Bella? Con nulla! Sarebbe stato molto più utile fare un paragone fra la terapia Di Bella e la terapia convenzionale». Come si vede, la progettazione della sperimentazione è stata considerata di basso livello, in quanto mancante delle 2 caratteristiche fondamentali che danno evidenza scientifica ad uno studio, e cioè la randomizzazione ed il gruppo di controllo.
La smentita definitiva dei risultati cui giunse tale sperimentazione è ulteriormente confermata dalle sempre più ampie e crescenti documentazioni, reperibili sulla letteratura, sull’efficacia antitumorale di quegli stessi principi attivi, utilizzati nel MDB (somatostatina, retinoidi, vitamina D3, Melatonina, ecc.), di cui la
sperimentazione aveva dichiarato l’inefficacia.
Tra le numerose e gravi anomalie rilevate:
progressivamente affinato e integrato dal medico e scienziato Prof. Luigi Di Bella, laureato anche in chimica e farmacia e docente universitario di fisiologia generale, fisiologia umana e biochimica, preferendolo alla chemioterapia. La tollerabilità della terapia e i positivi risultati in termini di sopravvivenza e di qualità di vita, portarono ad una sempre maggiore diffusione del MDB ( praticato da migliaia di pazienti), creando un grave contenzioso tra opinione pubblica e istituzioni sanitarie, che cercarono di eludere la sempre più pressante richiesta di erogazione gratuita del MDB con una sperimentazione di fase II, la cui progettazione, conduzione e conclusione, furono causa di aspre critiche e sconcerto, non solo nell’opinione pubblica, ma anche in ampi settori politici, medici e dell’informazione, fino a portare ad indagini della magistratura. Le anomalie di tale sperimentazione furono anche oggetto di oltre cinquanta interrogazioni parlamentari, pubblicate sulla Gazzetta Ufficiale del Parlamento Italiano. Indipendentemente dalle numerose e gravi anomalie documentate, e rilevate anche dalla letteratura internazionale .L’Istituto Superiore di Sanità italiano , responsabile della sperimentazione , ha sostenuto che: «E’ stato necessario effettuare uno studio senza gruppo di controllo perché, nella situazione esistente dall’inizio del 1998, ciò non era concepibile…».
Non era concepibile fare il tipo di studio che viene fatto in tutto il mondo, ed anche in Italia, correntemente (ma non per il MDB!?).
Questa argomentazione non tiene: infatti, quando la sperimentazione del MDB. è stata pubblicata sul British Medical Journal, l’editorialista Marcus Muller (fatto del tutto inusuale) ha criticato aspramente la progettazione dello studio, sostenendo che : «Gli autori (della sperimentazione) affermano anche che non avrebbero potuto condurre uno studio clinico randomizzato per ragioni etiche, ma queste ragioni non sono chiare. In realtà, c’è chi può sostenere che proprio il livello inferiore di progettazione dello studio è anti-etico».
Ed a proposito della mancanza del gruppo di controllo, ecco cosa afferma in una lettera al B.M.J. il ricercatore Rey M.D.: «Con cosa è stata comparata la terapia Di Bella? Con nulla! Sarebbe stato molto più utile fare un paragone fra la terapia Di Bella e la terapia convenzionale». Come si vede, la progettazione della sperimentazione è stata considerata di basso livello, in quanto mancante delle 2 caratteristiche fondamentali che danno evidenza scientifica ad uno studio, e cioè la randomizzazione ed il gruppo di controllo.
La smentita definitiva dei risultati cui giunse tale sperimentazione è ulteriormente confermata dalle sempre più ampie e crescenti documentazioni, reperibili sulla letteratura, sull’efficacia antitumorale di quegli stessi principi attivi, utilizzati nel MDB (somatostatina, retinoidi, vitamina D3, Melatonina, ecc.), di cui la
sperimentazione aveva dichiarato l’inefficacia.
Tra le numerose e gravi anomalie rilevate:
- l’erogazione di farmaci scaduti a 1048 pazienti [documentata e verbalizzata dai NAS (forze dell’ordine che in Italia hanno funzioni di controllo e verifica nella sanità)].
- Fu accertata altresì l’imperfetta preparazione dei farmaci (non rispettarono le indicazioni del Prof. Di Bella);
- la presenza di acetone, sostanza notoriamente tossica e cancerogena, in un farmaco della sperimentazione;
- la somministrazione di solo 4 dei sette farmaci previsti nel MDB;
- il mancato uso del temporizzatore per l’iniezione di somatostatina in un numero rilevante di pazienti. Ciò ha vanificando l’efficacia della somatostatina, che avendo un’emivita di 3 minuti, ha un’ efficacia strettamente condizionata a tempi di erogazione di almeno 8-10 ore con un temporizzatore;
- Furono scelti, per una terapia biologica, tempi e criteri di valutazione propri delle terapie citolitiche, disattendendo inoltre i criteri del National Cancer Institute (NCI) relativamente alla progettazione e obiettivi delle sperimentazioni cliniche. Il NCI ritiene infatti le progettazioni più affidabili quelle in doppio cieco e con gruppo di controllo, subito dopo quelle con gruppo di controllo, e a livello infimo la raccolta di casi clinici, quest’ultima, la meno scientificamente significativa, fu adottata per la sperimentazione del MDB. Relativamente agli obiettivi, quello primario per il NCI è la sopravvivenza, subito dopo la qualità di vita, infine le dimensioni della neoplasia. Quest’ultimo obiettivo, il meno affidabile, fu scelto per la sperimentazione.
- I criteri di arruolamento dei pazienti furono antitetici a quelli dichiarati e verbalizzati dal Prof Di Bella al Ministero della Sanità, in commissione oncologica, in sede di programmazione della sperimentazione. Il Prof Di Bella dichiarò che il suo metodo dava risultati direttamente proporzionali alla precocità di applicazione, e inversamente al numero e intensità dei cicli chemioradioterapici effettuati. Furono arruolati per la sperimentazione quasi interamente pazienti in condizioni critiche e/o terminali ripetutamente chemio- radiotrattati.
I responsabili della sperimentazione furono processati, non furono perseguiti perché si ritenne che le numerose e gravi anomalie, non smentite peraltro dalla magistratura, non fossero dovute a dolo, ma ad una messa a punto affrettata della sperimentazione, sotto la spinta dell’opinione pubblica. Permane e non fu smentito dalla magistratura, il dato di fatto delle numerose e gravi anomalie che hanno totalmente destituito di 49significato scientifico, e indicazioni cliniche, la sperimentazione. Esse furono denunciate oltre che dalle 50 interrogazioni parlamentari,(riportate sulla Gazzetta Ufficiale della Repubblica Italiana ) da una parte rilevante della stampa edell’informazione radiotelevisiva italiane. Esse sono dettagliatamente e ampiamente documentate nel volume di Vincenzo Brancatisano “Un po’ di verità sulla cura Di Bella“.Ed Trawel Factory .1999 .L’analisi dell’intera progettazione, conduzione e conclusione della sperimentazione supportata da documenti originali, verbali ministeriali, riscontri e verifiche è pubblicata nella monografia “Il Metodo Di Bella”, Mattioli Editore 3° Edizione 2005 del sottoscritto. Da questa documentazione la sperimentazione risulta totalmente delegittimata.
Ho ritenuto necessaria questa premessa e mi sono dilungato sia perché mi si potrebbe obiettare che la sperimentazione in Italia del 1998 ha dichiarato l’inefficacia del MDB , che per spiegare le ragioni che hanno portato in Italia al primo studio clinico osservazionale retrospettivo spontaneo, unicamente nato da iniziativa della gente, per contestare i risultati della sperimentazione e rivendicare la libertà di cura. In Italia migliaia di ammalati hanno fatto, e molti continuano anche oggi a fare, ricorso alla magistratura per ottenere l’erogazione gratuita del MDB. Dopo la sperimentazione, oltre duemila sentenze hanno condannato il Servizio Sanitario Italiano ad erogare il MDB, in base a perizie giurate che hanno certificato i positivi effetti della terapia in pazienti in cui la chemioterapiae/o anticorpi monoclonali si erano rivelati inefficaci.
Riteniamo utile segnalare il dato, oltre che per la rilevanza sociale, e l’assoluta mancanza di precedenti, anche per la notevole mole di dati scientifici e clinici emersi.
Riportiamo qui i dati di 124 pazienti oncologici esaminati da tre medici, in veste di “Consulenti Tecnici d’Ufficio” dalla Procura di Lecce, mentre altri 104 sono già presenti in letteratura e riportati su riviste recensite da Med-Line. Altri casi, suddivisi in gruppi omogenei per patologia, verranno pubblicati appena terminata l’elaborazione statistica ancora in corso.( www.metododibella.org)
Dopo la sperimentazione, oltre che per il “ricorso amministrativo”, volto a ottenere l’erogazione gratuita dei farmaci, molti ammalati inviarono spontaneamente la loro documentazione clinica alla Procura di Lecce, che essendo stata la prima a condannare il SSN ad erogare il MDB, era divenuta un punto di riferimento.
I consulenti tecnici di ufficio suddivisero così i casi:
A) pazienti che fecero ricorso alla Procura di Lecce per ottenere la cura;
B) ammalati che, indignati per le palesi e gravi anomalie della sperimentazione, di propria iniziativa avevano inviato a detta Procura le cartelle cliniche per
documentare i positivi effetti ottenuti col MDB.
La differenza tra i due gruppi consisteva soprattutto nel fatto che il gruppo A, in grande maggioranza, era costituito da pazienti che ricorrevano al MDB dopo il fallimento della chemioterapia, mentre nel secondo gruppo, tranne qualche eccezione, i pazienti avevano scelto il MDB come terapia di prima linea. Questo spiega l’opportunità, per una valutazione realistica e attendibile del MDB, di valutare solo il gruppo B, non inquinato da pregressa chemio.
Nella relazione della sentenza del Tribunale di Lecce viene indicato un periodo di osservazione pluriennale.
Dei 126 casi due sono deceduti in corso di trattamento, 124 furono monitorati per oltre 3 anni con percentuale significativa di pazienti osservati oltre i 5 anni.
L’analisi delle cartelle cliniche e delle relazioni dei medici curanti dei 124 del gruppo B ha fornito i seguenti dati:
Riportiamo qui i dati di 124 pazienti oncologici esaminati da tre medici, in veste di “Consulenti Tecnici d’Ufficio” dalla Procura di Lecce, mentre altri 104 sono già presenti in letteratura e riportati su riviste recensite da Med-Line. Altri casi, suddivisi in gruppi omogenei per patologia, verranno pubblicati appena terminata l’elaborazione statistica ancora in corso.( www.metododibella.org)
Dopo la sperimentazione, oltre che per il “ricorso amministrativo”, volto a ottenere l’erogazione gratuita dei farmaci, molti ammalati inviarono spontaneamente la loro documentazione clinica alla Procura di Lecce, che essendo stata la prima a condannare il SSN ad erogare il MDB, era divenuta un punto di riferimento.
I consulenti tecnici di ufficio suddivisero così i casi:
A) pazienti che fecero ricorso alla Procura di Lecce per ottenere la cura;
B) ammalati che, indignati per le palesi e gravi anomalie della sperimentazione, di propria iniziativa avevano inviato a detta Procura le cartelle cliniche per
documentare i positivi effetti ottenuti col MDB.
La differenza tra i due gruppi consisteva soprattutto nel fatto che il gruppo A, in grande maggioranza, era costituito da pazienti che ricorrevano al MDB dopo il fallimento della chemioterapia, mentre nel secondo gruppo, tranne qualche eccezione, i pazienti avevano scelto il MDB come terapia di prima linea. Questo spiega l’opportunità, per una valutazione realistica e attendibile del MDB, di valutare solo il gruppo B, non inquinato da pregressa chemio.
Nella relazione della sentenza del Tribunale di Lecce viene indicato un periodo di osservazione pluriennale.
Dei 126 casi due sono deceduti in corso di trattamento, 124 furono monitorati per oltre 3 anni con percentuale significativa di pazienti osservati oltre i 5 anni.
L’analisi delle cartelle cliniche e delle relazioni dei medici curanti dei 124 del gruppo B ha fornito i seguenti dati:
| totale | remissione | regressione | stabilità | progressione |
| 124 | 12 | 58 | 37 | 17 |
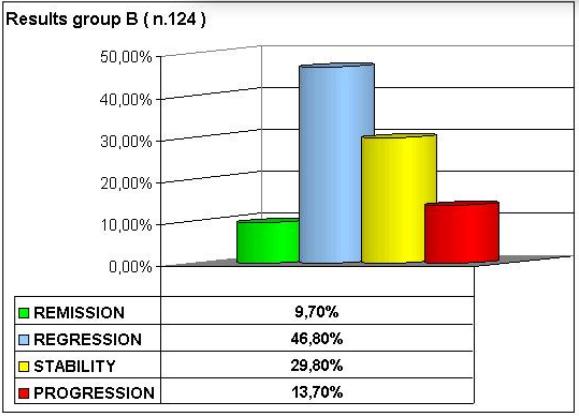
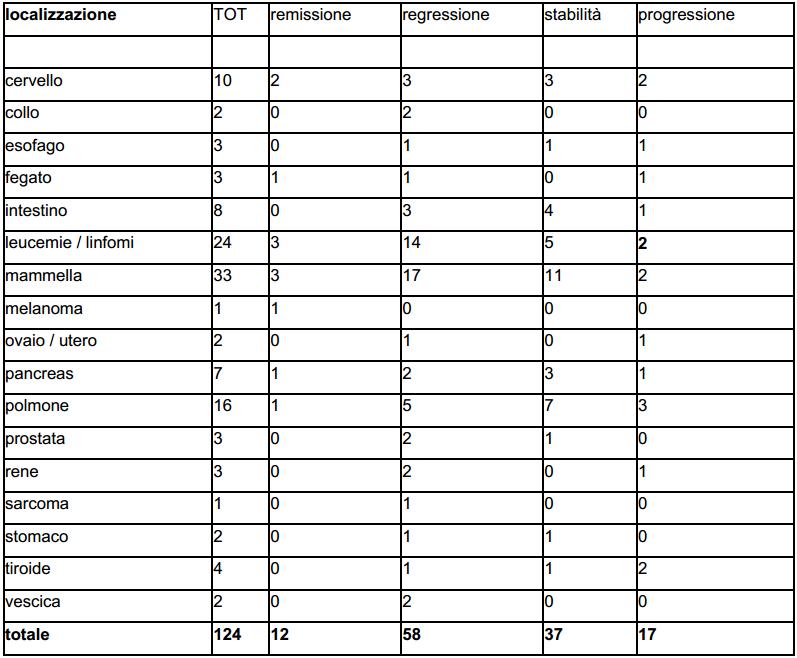
In particolare:
Leucemie - Linfomi
| totale | remissione | regressione | stabilità | progressione |
| 24 | 3 | 14 | 5 | 2 |
- Note: (1 caso, dei 2 in progressione, non usava melatonina adeguata. )
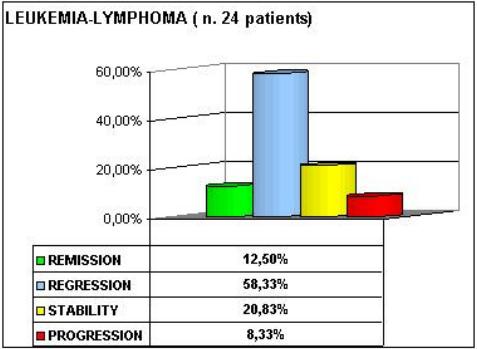
Mammella
| totale | remissione | regressione | stabilità | progressione |
| 33 | 3 | 17 | 11 | 2 |
- Note: (i 2 casi in progressione erano precedentemente stati sottoposti a Chemio e plurimetastatizzati)
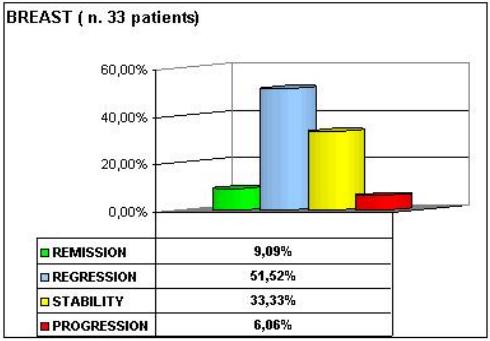
Pancreas
| totale | remissione | regressione | stabilità | progressione |
| 7 | 1 | 2 | 3 | 1 |
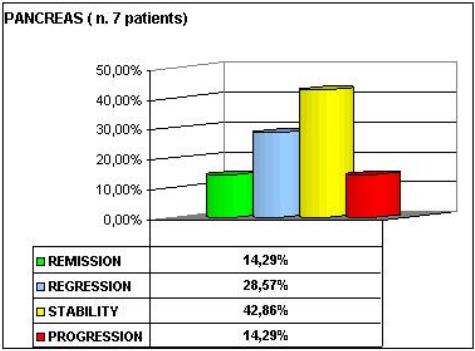
Cervello
| totale | remissione | regressione | stabilità | progressione |
| 10 | 2 | 3 | 3 | 2 |
- Note: (i 2 casi in progressione erano: 1 precedentemente sottoposto a Chirurgia/ CH / RT , l'altro con documentazione incompleta)
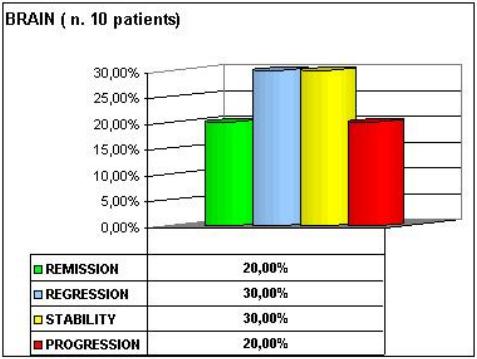
Polmone
| totale | remissione | regressione | stabilità | progressione |
| 16 | 1 | 5 | 7 | 3 |
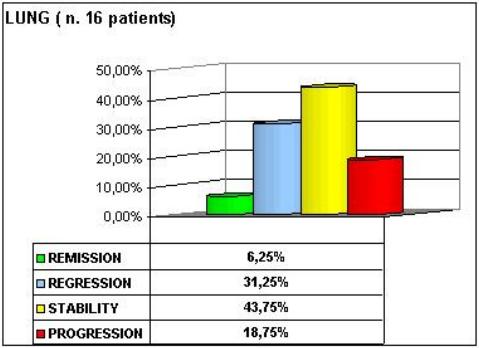
Inoltre, è stato possibile misurare anche il dato sulla qualità di vita dei 124 pazienti:
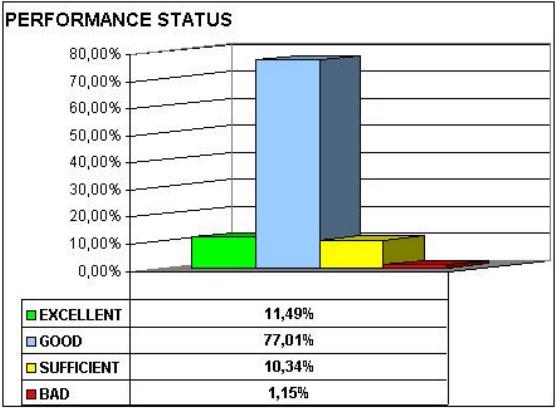
Le concordi conclusioni e dichiarazioni dei 3 periti del Tribunale di Lecce, relativamente alla totalità dei casi esaurientemente documentati, hanno portato ad una chiara dichiarazione di efficacia e tollerabilità del MDB.
In base a queste perizie giurate il Magistrato ha dichiarato che:
1) Dalla copiosa documentazione clinica acquisita agli atti del processo, emerge il dato incontrovertibile che moltissimi pazienti, al di fuori della sperimentazione, con la MDB hanno ottenuto risultati positivi, non soltanto a livello di miglioramento della qualità di vita, ma anche di blocco o di regressione del male”
2) Le dichiarazioni testimoniali di medici e di ammalati o di parenti degli ammalati sul recupero totale o parziale della salute a seguito dell’utile ricorso alla terapia Di Bella[...]. Dalla relazione peritale dei consulenti tecnici di ufficio […] tutti (i pazienti) hanno avuto miglioramenti a livello della qualità di vita […] e stanno avendo risultati positivi per via del notevole prolungamento dei tempi di sopravvivenza rispetto alla prognosi iniziale infausta a brevissima scadenza (poche settimane di vita).
In conclusione la MDB sembra aver determinato un miglioramento della qualità di vita nella maggior parte dei casi trattati.
Non si sono mai rilevati effetti collaterali legati alla MDB.
Non sfugge al giudicante il contrasto tra le risultanze dei periti di ufficio e le conclusioni della sperimentazione ufficiale […].Il miglioramento delle condizioni di vita degli ammalati neoplastici, conseguente all’assunzione dei farmaci della MDB, è un dato più che sufficiente a giustificare la somministrazione di detta terapia, e ciò in funzione della tutela del bene giuridico assoluto della salute, cui fa riferimento l’art. 32 della Costituzione, che altrimenti risulterebbe gravemente ed irreparabilmente pregiudicato”.
Queste conclusioni di un magistrato, basate su relazioni cliniche, testimonianze e dati documentali validati da perizie mediche giurate, sono scientificamente significative, meritano attenta e profonda esame, contrastano radicalmente e smentiscono le conclusioni di quella sperimentazione le cui anomalie e la totale inaffidabilità di impostazione, conduzione e conclusione, sono ormai documentate e note.
Aggiungendo a questi 124 casi certificati dai periti del tribunale di Lecce i seguenti dati, si ha la dimensione complessiva della statistica, che comprende:
54 casi pubblicati sulla rivista dell’associazione di medici che in Italia applicano il MDB :
- 23 casi di linfomi NH
- 26 casi di carcinomi della mammella
- 2 carcinomi polmonari a piccole cellule
- 1 sarcoma osteogenico
- 1 mesotelioma pleurico
- 1 neoplasia cerebrale
per un totale di 54 casi
relazioni sulle risultanze del MDB presentate al 1° e 2° Congresso nazionale MDB e al Congresso sulla Terapia biologica delle malattie neoplastiche e degenerative :
222 casi presentati al 1° Congresso nazionale sul MDB Bologna 2004
Un centinaio circa di casi tra carcinomi anaplastici, tumori della mammella, polmonari, Linfomi NH, osservati per un periodo oscillante fra i tre e i sette anni
relazioni sulle risultanze del MDB presentate al 1° e 2° Congresso nazionale MDB e al Congresso sulla Terapia biologica delle malattie neoplastiche e degenerative :
222 casi presentati al 1° Congresso nazionale sul MDB Bologna 2004
Un centinaio circa di casi tra carcinomi anaplastici, tumori della mammella, polmonari, Linfomi NH, osservati per un periodo oscillante fra i tre e i sette anni
- 1 caso di mieloma multiplo
- 1 astrocitoma
- 1 adenocarcinoma mucinoso dell’ovaio
- 1 adenocarcinoma metastatico del sigma
- 12 casi di mesoteliomi pleurici
- 1 Adenocarcinoma colangiocellulare
- 6 Linfomi NH
- 2 Ca metastatici della mammella
- 1 Epatocarcinoma
- 1 carcinoma ovarico
- 2 adenocarcinomi del pancreas
- 76 Casi di tumori polmonari
- 1 .Adenomatosi multipla del fegato
- 2 Carcinomi polmonari
- 1 sarcoma
- 1 leiomiosarcoma
- 2 Carcinomi del colon retto
- 1 carcinoma laringeo
- 1 liposarcoma pleomorfo
- 1 Mesotelioma pleurico
- 1 Linfoma gastrico
- 1 adenocarcinoma polmonare non a piccole cellule
- 1 carcinoma epidermide polmonare
- 3 carcinomi gastrici
- 1 sarcoma
- 1 carcinoma epato pancreatico
- 1 carcinoma indifferenziato pleuropolmonare
- 1 ca mammella
Totale: 222 casi
120 casi presentati al 2° Congresso nazionale sul MDB Milano 2005
120 casi presentati al 2° Congresso nazionale sul MDB Milano 2005
- 28 carcinomi polmonari non a piccole cellule
- 46 ca polmonari a piccole cellule
- 2 linfomi NH
- 1 leucemia linfatica cronica
- 17 Carcinomi pancreatici
- 6 Carcinomi mammari
- 1 Ca anaplastico del polmone
- 1 linfoma NH
- 2 linfomi NH
- 4 carcinomi del pancreas esocrino
- 1 sarcoma
- 2 tumori metastatici della mammella
- 1 mieloma multiplo
- 1 linfoma NH
- 1 carcinoma mammario con disseminazioni polmonari
- 4 linfomi NH
- 2 tumori metastatici della mammella
- 1 epatocarcinoma
- 2 sarcomi
Totale: 120 casi
12 casi presentati Congresso del 16 gennaio 2010 presso la Repubblica di S Marino sulla” Terapia biologica delle malattie neoplastiche e degenerative”
12 casi presentati Congresso del 16 gennaio 2010 presso la Repubblica di S Marino sulla” Terapia biologica delle malattie neoplastiche e degenerative”
- 4 leucemie linfoblastiche
- 1 neuroblastoma
- 1 carcinoma renale
- 2 ca mammella
- 1 carcinoma tiroideo
- 1 epatocarcinoma
- 1 leiomiosarcoma
- 1 Ca testicolo
Totale: 12 casi
Gli atti di questi congressi sono stati pubblicati e reperibili sul portale della Fondazione Di Bella www.metododibella.org
103 casi già pubblicati su riviste recensite da Med-Line
Gli atti di questi congressi sono stati pubblicati e reperibili sul portale della Fondazione Di Bella www.metododibella.org
103 casi già pubblicati su riviste recensite da Med-Line
- 20 casi di ‘linfomi NHL pubblicati su Cancer Biotherapy
- 4 casi di leucemia linfoblastica “” “”
- 1 caso di NHL Am. J. Therapy
- 1 caso di NHL Am. J. Therapy
- Un caso di carcinoma della mammella pubblicato su NEL
- 1 caso di carcinoma dell’esofago p pubblicato su NEL
- Un caso di neuroblastoma pubblicato su NEL
- 74 casi di carcinomi polmonari pubblicti da Cancer Biotherapy,
Per un totale di 103 casi
Dimensioni della statistica complessiva di pazienti curati con MDB: 635 casi
Conclusioni
Si possono evidenziare alcuni dati che, anche se con frequenza, tempi, modalità e intensità diversa, sono comunemente riscontrabili o relativamente frequenti nei pazienti neoplastici trattati con MDB:
- Incremento generalizzato dell’aspettativa di vita, miglioramento della sua qualità, diminuzione della frequenza, numero ed entità delle disseminazione neoplastiche, maggior contenimento e controllo della progressione sia delle lesioni primitive che secondarie.
- Riduzione dell’attività metabolica delle popolazioni cellulari neoplastiche,evidenziata dalla PET associata a contenimento e/o eliminazione delle sindromi paraneoplastiche neurologiche, vascolari, flogistiche ecc…
- Maggior controllo delle lesioni secondarie osteolitiche, migliore capacità di riportare a livelli fisiologici il metabolismo, trofismo e funzionalità del tessuto osteocatolagineo,.degli epiteli, della matrice extacellulare.
- Miglioramento della cenestesi, dell’appetito, della funzionalità neuromuscolare, del trofismi di parenchimi, tessuti e degli endoteli con regolazione fisiologica della permeabilità degli stessi e conseguentemente degli scambi emotissutali.
- Detersione e riepitelizzazione di focolai misti flogistico-neoplastici.
- Miglioramento della micro e macrocircolazione viscerale.
- Miglioramento psicofisico, osservabile non di rado anche in soggetti gravemente sofferenti e in condizioni critiche.
- Diminuzione per tempi più o meno protratti della velocità di progressione con stabilizzazione di patologie neoplastiche avviate a evidente e rapida progressione prima dell’inizio della multiterapia MDB.
- Possibile controllo e/o attenuazione del dolore con diminuzione del dosaggio di analgesici o loro sospensione.
- Spostamento verso parametri fisiologici dell’aggregazione piastrinica ,e tollerabilità delle minimali e continuative dosi di ciclofosfamide o oncocarbide per l’effetto mieloprotettivo della MLT complessata e del composto dei retinoidi MDB
- Miglioramento dell’immunità con diminuzione dell’insorgenza di processi flogistici, più agevolmente dominabili.
- Rapidità ed intensità di risposta soggettiva e obiettiva al MDB inversamente proporzionale:
- all’ intensità e al numero dei cicli chemioterapici effettuati;
- all’intervallo di tempo intercorso dall’insorgenza del tumore;
- ai cicli radioterapici effettuati (ad eccezione della radioterapia stereotassica).
- Alle condizioni cliniche del paziente all’inizio del MDB.
- Se il MDB è somministrato contemporaneamente alla radioterapia esercita un effetto sia radioprotettivo che radiosensibilizzante.
- Alla stabilizzazione di neoplasie in progressione prima del trattamento con MDB, segue frequentemente una convivenza col tumore con qualità di vita accettabile che può consentire la ripresa parziale o totale dell’attività lavorativa. Questa sorta di omeostasi paziente-tumore si può non raramente osservare per uno o più anni, anche in pazienti che sembravano rapidamente avviati all’exitus, se le condizioni non sono gravemente e irrimediabilmente compromesse da insufficienza funzionale di organi vitali più frequentemente se non sono stati effettuati ripetuti e pesanti cicli chemioterapici.
- Se il MDB è applicato contemporaneamente ai protocolli chemioterapici,ne potenzia notevolmente l’azione antiblastica i, riducendone sensibilmente la tossicità.
- Quando si ha una progressione questa ha un andamente molto più lento e graduale rispetto ad analoghe situazione in pazienti chemio e/o radiotrattati.
- Nei rari casi in cui è stato possibile iniziare il MDB almeno 20 – 30 giorni prima della exeresi chirurgica di neoplasie, non si sono notate recidive.Nei tumori operabili i migliori risultati si ottengono facendo precedere, accompagnare e seguire dal MDB l’intervento, con netta diminuzione della possibile disseminazione intraoperatoria di cellule neoplasiche conseguente all’apertura di vasi sanguigni, linfatici e delle naturali fasce di contenimento dell’espansione neoplastica, oltre che alla potente e documentata attivazione chirurgica dell’angiogenes neoplastica.L’inibizione dell’angiogenesi si ottiene attraverso la regolazione negativa dei molteplici fattori che intervengono nel complesso meccanismo dell’angiogenesi tumorale.
- Dopo un periodo più o meno prolungato di stabilità, molte volte si può verificare una progressione, ma in un numero di casi vario per patologia tumorale e stadio, una lenta e graduale riduzione volumetrica e numerica delle localizzazioni primitive e secondarie e, a seconda delle caratteristiche istologiche, una completa remissione.
- Maggiore evidenza , rapidità e frequenza di risposte positive in termini di remissione miglioramento della qualità di vita,risposte obiettive, si registrano nei linfomi Hodgkin e NH, nelle LLC, nei tumori neuroendocrini, nei carcinomi tiroidei soprattutto midollari e papillari,nei neuroblastomi,chemodectomi,tumori glomici carcinomi della mammella, nei sarcomi nei carcinomi degli epiteli aerodigestivi superiori, degli annessi ghiandolari esocrini, e nei carcinomi prostatici , vescica.
- Una risposta inferiore in termini di frequenza di stabilizzazione, remissione, incremento dell’aspettativa di vita, (ma per mediana di sopravvivenza, qualità di vita e tollerabilità della terapia nettamente e indubbiamente superiore ai comuni trattamenti chemioterapici) si verifica nei tumori polmonari NSCLC, mentre rispondono meglio quelli a piccole cellule,nei carcinomi esocrini del pancreas ,negli epato carcinomi, carcinomi del colon retto, dello stomaco, del rene, dell’utero, ovaio, mesoteliomi.
- Nei melanomi, glioblastomi, colangio carcinomi,nei tumori metastatici del rene a cellule chiare, le mediane di sopravvivenza sono inferiori alle precedenti, così come le remissioni e il controllo delle estensioni locoregionali e/o disseminazioni a distanza, anche se in queste forme, come nelle precedenti, la mediana di sopravvivenza, relative ad ogni stadio,la qualità di vita e tollerabilità della terapia sono ampiamente superiori ai comuni trattamenti chemioterapici. Essi in queste patologie non consentono alcun apprezzabile risultato, Il MDB ha dimostrato rispetto alla chemioterapia un netto miglioramento della qualità e della durata della vita, senza alcuno dei rilevanti effetti tossici della chemio.
- In tutti i casi, con rare eccezioni, si assiste ad un evidente miglioramento delle qualità di vita, dato comune a tutte le patologie neoplastiche trattate con MDB, così come si evidenzia una assenza di tossicità rilevante al MDB ad eccezione di una certa sonnolenza dovuta alla MLT e di transitoria tossicità di 1° o 2°relativa a nausea o diarrea. Questi sintomi non hanno mai carattere di gravità, e sono temporanei sia per i meccanismi di compenso fisiologici, sia per la possibilità di intervenire con qualche accorgimento, come la somministrazione della MLT,prevalentemente nelle ore notture e 62della somatostatina almeno 3 ore dopo cena, portando l’infusione con temporizzatore a 10 ore.
- Molto più difficilmente con MDB si verificano le situazioni di cahessia neoplastica dei pazienti terminali, ma fino all’exitus non raramente si risparmiano all' ammalato drammatici decadimenti nè gravi sofferenze.
Il numero di casi clinici monitorati, il periodo di osservazione, che in molti casi ha superato i cinque anni, e la grande varietà di istotipi neoplastici, consentono di trarre lcune conclusioni preliminari limitate alle mediane di sopravvivenza, alla qualità di vita, e alla tollerabilità del MDB.Consideriamo preliminari questi dati , anche se documentati e certificati ,in quanto derivanti da uno studio osservazionale retrospettivo , e perché la suddivisione in patologie omogenee, e l’elaborazione statistica delle risposte obiettive delle singole patologie è ancora in corso, anche se di prossima pubblicazione . Ricordiamo comunque che al primo posto degli obiettivi di uno studio clinico, il National Cancer Institute pone la sopravvivenza dell’ammalato neoplastico , e al secondo la qualità di vita. Pertanto questi dati non sono privi di dignità e rilevanza scientifica.
Dalla revisione della statistica complessiva emerge un’ evidente uniformità di dati relativi alla mediana di sopravvivenza e alla qualità di vita. Sono sostanzialmente sovrapponibili quelli dei 3 congressi e delle pubblicazioni sulla rivista italiana ( non recensita da Med Line ), delle riviste internazionali recensite su Med Line, e dei periti del Tribunale di Lecce .
In tutte le neoplasie trattate con MDB, anche se con rilevanti differenze tra di esse, si osserva un netto incremento dell’aspettativa di vita e un miglioramento della sua qualità, in assenza di rilevante tossicità rispetto ai dati della letteratura relativi alle stesse neoplasie e allo stesso stadio.
Emerge anche chiaramente che la risposta al MDB è direttamente proporzionale alla precocità del trattamento, e inversamente proporzionale al numero e all’intensità dei cicli chemio-radioterapici effettuati.
Se il MDB precede gli interventi le recidive sono molto più rare rispetto ai dati della letteratura.
Riteniamo che il confronto delle documentate basi scientifiche, della logica lineare e matematica del razionale del MDB, dei significativi risulati , con i gravi e noti limiti delle attuali terapie antitumorali, possa indurre ad un maggior interesse sulle prospettive aperte dal MDB. Il tumore è deviazione dalla vita normale, che il MDB corregge, assecondando ed esaltando le reazioni vitali.
Il Metodo Di Bella , non è pertanto ”alternativo”, nell’accezione comune del termine, ma rappresenta l’integrazione razionale e la convergenza delle conoscenze medico-scientifiche definitivamente acquisite, e delle emergenti evidenze scientifiche, in una clinica affrancata da inquinamenti politico-finanziari.
Dalla revisione della statistica complessiva emerge un’ evidente uniformità di dati relativi alla mediana di sopravvivenza e alla qualità di vita. Sono sostanzialmente sovrapponibili quelli dei 3 congressi e delle pubblicazioni sulla rivista italiana ( non recensita da Med Line ), delle riviste internazionali recensite su Med Line, e dei periti del Tribunale di Lecce .
In tutte le neoplasie trattate con MDB, anche se con rilevanti differenze tra di esse, si osserva un netto incremento dell’aspettativa di vita e un miglioramento della sua qualità, in assenza di rilevante tossicità rispetto ai dati della letteratura relativi alle stesse neoplasie e allo stesso stadio.
Emerge anche chiaramente che la risposta al MDB è direttamente proporzionale alla precocità del trattamento, e inversamente proporzionale al numero e all’intensità dei cicli chemio-radioterapici effettuati.
Se il MDB precede gli interventi le recidive sono molto più rare rispetto ai dati della letteratura.
Riteniamo che il confronto delle documentate basi scientifiche, della logica lineare e matematica del razionale del MDB, dei significativi risulati , con i gravi e noti limiti delle attuali terapie antitumorali, possa indurre ad un maggior interesse sulle prospettive aperte dal MDB. Il tumore è deviazione dalla vita normale, che il MDB corregge, assecondando ed esaltando le reazioni vitali.
Il Metodo Di Bella , non è pertanto ”alternativo”, nell’accezione comune del termine, ma rappresenta l’integrazione razionale e la convergenza delle conoscenze medico-scientifiche definitivamente acquisite, e delle emergenti evidenze scientifiche, in una clinica affrancata da inquinamenti politico-finanziari.